Ingrédients actifs : Emtricitabine, Ténofovir disoproxil
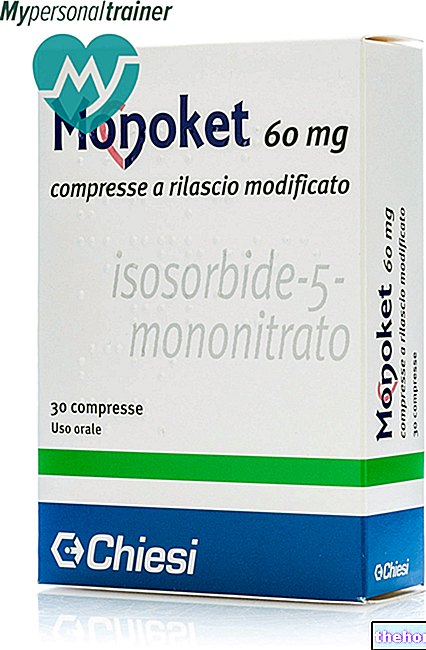
Truvada 200 mg / 245 mg comprimés pelliculés
Pourquoi Truvada est-il utilisé ? Pourquoi est-ce?
Truvada est un traitement de l'infection par le virus de l'immunodéficience humaine (VIH) chez les adultes âgés de 18 ans et plus.
Truvada contient deux substances actives, l'emtricitabine et le ténofovir disoproxil. Les deux principes actifs sont des médicaments antirétroviraux utilisés pour traiter l'infection par le VIH. L'emtricitabine est un inhibiteur nucléosidique de la transcriptase inverse et le ténofovir est un inhibiteur nucléotidique de la transcriptase inverse. Cependant, ils sont génériquement appelés INTI et agissent en interférant avec l'activité normale d'une enzyme ( transcriptase inverse) qui est essentielle à la reproduction du virus. Truvada doit toujours être utilisé en association avec d'autres médicaments pour traiter l'infection par le VIH. Truvada peut être administré en remplacement de l'emtricitabine et du ténofovir disoproxil utilisés séparément aux mêmes doses.
Ce médicament n'est pas un remède contre l'infection par le VIH. Vous pouvez toujours développer des infections ou d'autres maladies associées à l'infection par le VIH pendant que vous prenez Truvada. Vous pouvez toujours transmettre le VIH pendant que vous prenez ce médicament, bien que le risque soit réduit par l'effet du traitement antirétroviral. Discutez avec votre médecin des précautions nécessaires pour éviter de transmettre l'infection à d'autres personnes.
Contre-indications Quand Truvada ne doit pas être utilisé
Ne prenez pas Truvada
- Si vous êtes allergique à l'emtricitabine, au ténofovir, au fumarate de ténofovir disoproxil ou à l'un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Si cela vous concerne, informez-en immédiatement votre médecin.
Précautions d'emploi Quelles sont les informations à connaître avant de prendre Truvada
- Informez votre médecin si vous avez eu une maladie rénale ou si les tests ont montré des problèmes rénaux. Truvada peut affecter les reins. Avant de commencer le traitement, votre médecin peut vous prescrire des analyses de sang pour évaluer la fonction rénale correcte. Votre médecin peut également demander des analyses de sang pendant le traitement pour surveiller vos reins et peut vous conseiller de prendre les comprimés moins fréquemment. Truvada n'est pas recommandé si vous souffrez d'une maladie rénale grave ou si vous êtes sous hémodialyse. Truvada ne doit pas être pris avec d'autres médicaments pouvant endommager les reins (voir Autres médicaments et Truvada). Si cela est inévitable, votre médecin surveillera votre fonction rénale une fois par semaine.
- Informez votre médecin si vous avez plus de 65 ans. Truvada n'a pas été étudié chez les patients de plus de 65 ans. Si vous avez plus de cet âge et qu'on vous a prescrit Truvada, votre médecin vous surveillera étroitement.
- Informez votre médecin si vous avez déjà eu des problèmes de foie, y compris une hépatite. Les patients atteints de problèmes hépatiques, y compris l'hépatite B ou C chronique qui sont traités avec des antirétroviraux, ont un risque plus élevé de complications hépatiques graves pouvant entraîner la mort.Si vous souffrez d'hépatite B, votre médecin examinera attentivement la meilleure option de traitement pour vous. Les deux principes actifs contenus dans Truvada ont une certaine activité contre le virus de l'hépatite B, bien que l'emtricitabine ne soit pas autorisée pour le traitement de l'hépatite B. Si vous avez eu une maladie du foie ou une hépatite B chronique, votre médecin peut vous prescrire des analyses de sang pour surveiller avec précision votre la fonction hépatique.
Autres précautions
Les thérapies antirétrovirales combinées (y compris Truvada) peuvent augmenter la glycémie, les taux de lipides sanguins (hyperlipémie), entraîner des modifications de la graisse corporelle et une résistance à l'insuline (voir rubrique 4, Effets indésirables éventuels).
Si vous êtes diabétique, en surpoids ou avez un taux de cholestérol élevé, veuillez en informer votre médecin.
Attention aux infections. Si vous êtes atteint d'un stade avancé du VIH (SIDA) et que vous avez une infection, vous pouvez développer des symptômes d'"infection et d'inflammation ou une aggravation des symptômes d'une infection existante lorsque vous commencez le traitement par Truvada. Ces symptômes peuvent indiquer que le système immunitaire de votre corps combat l'infection. Recherchez des signes d'inflammation ou d'infection peu de temps après avoir commencé à prendre Truvada.Si vous remarquez des signes d'inflammation ou d'infection, informez-en immédiatement votre médecin.
En plus des infections opportunistes, des troubles auto-immuns (une affection qui survient lorsque le système immunitaire attaque des tissus corporels sains) peuvent également survenir après que vous ayez commencé à prendre des médicaments pour traiter l'infection par le VIH. Des troubles auto-immuns peuvent survenir plusieurs mois après le début du traitement.Si vous remarquez des symptômes d'infection ou d'autres symptômes tels qu'une faiblesse musculaire, une faiblesse initiale des mains et des pieds remontant jusqu'au tronc, des palpitations, des tremblements ou une hyperactivité, informez votre médecin immédiatement pour demander le traitement nécessaire.
Problèmes osseux. Certains patients prenant un traitement antirétroviral combiné peuvent développer une maladie osseuse appelée ostéonécrose (mort du tissu osseux causée par un manque d'apport sanguin à l'os). Durée du traitement antirétroviral combiné, utilisation de corticostéroïdes, consommation d'alcool, immunosuppression sévère, un indice de masse corporelle plus élevé, entre autres, peut être l'un des nombreux facteurs de risque de développer cette maladie. Les signes d'ostéonécrose sont une raideur articulaire, des courbatures (en particulier dans les hanches, les genoux et les épaules) et des difficultés à bouger. Contactez votre médecin si vous remarquez l'un de ces symptômes.
Des problèmes osseux (entraînant parfois des fractures) peuvent également survenir en raison de lésions des cellules tubulaires des reins (voir rubrique 4, Effets indésirables éventuels).
Enfants et adolescents
- Truvada n'est pas indiqué chez les enfants et les adolescents de moins de 18 ans.
Interactions Quels médicaments ou aliments peuvent modifier l'effet de Truvada
Autres médicaments et Truvada
Vous ne devez pas prendre Truvada si vous prenez déjà d'autres médicaments contenant les composants de Truvada, de l'emtricitabine et du fumarate de ténofovir disoproxil, ou tout autre médicament antiviral contenant de la lamivudine ou de l'adéfovir dipivoxil.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Il est particulièrement important d'informer votre médecin si vous prenez d'autres médicaments pouvant nuire à vos reins. Ceux-ci inclus:
- aminosides (pour infection bactérienne)
- amphotéricine B (pour les infections fongiques)
- foscarnet (pour infection virale)
- ganciclovir (pour infection virale)
- pentamidine (pour les infections)
- vancomycine (pour infection bactérienne)
- interleukine-2 (pour traiter le cancer)
- cidofovir (pour infection virale)
- anti-inflammatoires non stéroïdiens (AINS, utilisés pour soulager les douleurs osseuses ou musculaires)
Autres médicaments contenant de la didanosine (pour l'infection par le VIH) : la prise de Truvada avec d'autres médicaments antiviraux contenant de la didanosine peut augmenter le taux de didanosine dans le sang et réduire le nombre de cellules CD4. Lorsque des médicaments contenant du fumarate de ténofovir disoproxil et de la didanosine sont pris ensemble, De rares cas d'inflammation du pancréas et d'acidose lactique (excès d'acide lactique dans le sang), entraînant parfois la mort.
N'arrêtez pas le traitement sans contacter votre médecin.
Truvada avec de la nourriture et des boissons
Truvada doit être pris avec de la nourriture.
Avertissements Il est important de savoir que :
La grossesse et l'allaitement
Si vous êtes enceinte ou si vous allaitez, si vous pensez être enceinte ou prévoyez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant de prendre ce médicament.
- Vous ne devez pas prendre Truvada pendant la grossesse à moins d'en avoir spécifiquement discuté avec votre médecin. Bien qu'il existe des données cliniques limitées sur l'utilisation de Truvada chez la femme enceinte, il n'est généralement pas utilisé sauf en cas de stricte nécessité.
- Si vous êtes une femme susceptible de tomber enceinte pendant le traitement par Truvada, vous devez utiliser une contraception efficace pour l'éviter.
- Si vous êtes enceinte ou envisagez de le devenir, demandez à votre médecin quels sont les avantages et les risques potentiels du traitement par Truvada pour vous et votre bébé.
Si vous avez déjà pris Truvada pendant votre grossesse, votre médecin peut vous prescrire régulièrement des analyses de sang et d'autres tests de diagnostic pour surveiller le développement du bébé. Chez les enfants dont les mères ont pris des INTI pendant la grossesse, le bénéfice de la protection contre l'infection par le VIH l'emportait sur le risque d'effets secondaires.
- N'allaitez pas pendant que vous prenez Truvada. La raison en est que l'ingrédient actif de ce médicament est excrété dans le lait maternel.
- Si vous êtes une femme infectée par le VIH, il est recommandé de ne pas allaiter, afin d'éviter de transmettre le virus du VIH au bébé par le lait.
Conduire et utiliser des machines
Truvada peut provoquer des étourdissements. Si vous vous sentez étourdi pendant que vous prenez Truvada, ne conduisez pas et n'utilisez pas d'outils ou de machines.
Truvada contient du lactose
Informez votre médecin si vous avez une « intolérance au lactose ou à d'autres sucres. Truvada contient du lactose monohydraté. Si vous savez que vous êtes intolérant au lactose ou si votre médecin vous a dit que vous avez une intolérance à certains sucres, contactez votre médecin avant de prendre ce médicament.
Dose, mode et heure d'administration Comment utiliser Truvada : Posologie
Prenez toujours ce médicament en suivant exactement les indications de votre médecin. En cas de doute, consultez votre médecin ou votre pharmacien.
La dose recommandée est :
- Adultes : un comprimé par jour à prendre avec de la nourriture.
Si vous avez des difficultés à avaler, vous pouvez utiliser le bout d'une cuillère pour écraser le comprimé, puis mélanger la poudre dans environ 100 ml (un demi-verre) d'eau, de jus d'orange ou de jus de raisin et boire immédiatement.
- Prenez toujours la dose recommandée par votre médecin. Cela permet de s'assurer que vos médicaments sont pleinement efficaces et de réduire le risque de développer une résistance au traitement. Ne modifiez pas votre dose à moins que votre médecin ne vous le dise.
- Si vous avez des problèmes rénaux, votre médecin peut vous demander de prendre Truvada moins fréquemment.
- Si votre médecin décide d'arrêter l'un des composants de Truvada ou de modifier la dose de Truvada, l'emtricitabine et/ou le ténofovir peuvent vous être administrés séparément au lieu de l'association médicamenteuse ou d'autres médicaments pour le traitement de l'infection par le VIH.
- Votre médecin vous prescrira Truvada avec d'autres médicaments antirétroviraux. Consultez les notices des autres antirétroviraux pour obtenir des conseils sur la prise de ces médicaments.
Surdosage Que faire si vous avez pris trop de Truvada
Si vous avez pris plus de Truvada que vous n'auriez dû
Si vous prenez accidentellement plus que la dose recommandée de Truvada, contactez votre médecin ou le centre d'urgence le plus proche. Emportez le flacon de comprimés avec vous afin de pouvoir décrire facilement ce que vous avez pris.
Si vous oubliez de prendre Truvada
Il est important que vous ne manquiez aucune dose de Truvada.
Si vous oubliez une dose de Truvada dans les 12 heures suivant l'heure habituelle de prise, prenez-la dès que possible, puis prenez la dose suivante à l'heure habituelle.
S'il est presque temps (moins de 12 heures) pour votre prochaine dose, sautez la dose oubliée. Attendez et prenez la dose suivante régulièrement. Ne prenez pas de dose double pour compenser un comprimé oublié.
Si vous vomissez dans l'heure qui suit la prise de Truvada, prenez un autre comprimé. Vous ne devez pas prendre un autre "comprimé si vous avez vomi plus d'une" heure après avoir pris Truvada.
Si vous arrêtez de prendre Truvada
- L'arrêt de Truvada peut réduire l'efficacité du traitement anti-VIH prescrit par votre médecin. Adressez-vous à votre médecin avant d'arrêter de prendre Truvada pour quelque raison que ce soit, en particulier si vous avez ressenti un effet indésirable ou si vous souffrez d'une autre maladie. Contactez votre médecin avant de recommencer à prendre les comprimés de Truvada.
- Si vous êtes infecté par le VIH et l'hépatite B, il est particulièrement important de ne pas arrêter de prendre Truvada sans avoir d'abord contacté votre médecin. Certains patients ont présenté une aggravation de leur hépatite, comme l'indiquent les symptômes ou les analyses de sang après l'arrêt de Truvada. Il peut être nécessaire de répéter les analyses de sang pendant plusieurs mois après l'arrêt du traitement. Chez certains patients atteints d'une maladie hépatique avancée ou d'une cirrhose, l'arrêt du traitement n'est pas recommandé car cela peut entraîner une aggravation de l'hépatite.
Signalez immédiatement à votre médecin tout symptôme nouveau ou inhabituel observé après l'arrêt du traitement, en particulier les symptômes normalement associés à une infection par l'hépatite B.
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou votre pharmacien.
Effets secondaires Quels sont les effets secondaires du Truvada
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, bien que tout le monde n'y soit pas sujet.
Contactez votre médecin pour l'un des effets secondaires suivants :
Effets secondaires graves possibles : contactez immédiatement votre médecin
L'effet indésirable suivant est rare (survient chez au maximum 1 patient sur 1 000) : acidose lactique (excès d'acide lactique dans le sang), effet indésirable grave pouvant être fatal. Les effets secondaires suivants peuvent être des signes d'acidose lactique :
- respiration sifflante
- somnolence
- sensation de nausées, vomissements et douleurs à l'estomac
Si vous pensez souffrir d'acidose lactique, contactez immédiatement votre médecin.
Autres effets secondaires graves possibles
Les effets indésirables suivants sont peu fréquents (survenant chez au maximum 1 patient sur 100) :
- douleur abdominale causée par une inflammation du pancréas
- gonflement du visage, des lèvres, de la langue ou de la gorge
Les effets indésirables suivants sont rares (survenant chez au maximum 1 patient sur 1 000) :
- foie gras
- peau et yeux jaunes, démangeaisons ou douleurs abdominales causées par une inflammation du foie
- inflammation des reins, urine abondante et soif, insuffisance rénale, lésions des cellules tubulaires rénales. Votre médecin peut vous prescrire des analyses de sang pour voir si vos reins fonctionnent correctement.
- ramollissement des os (avec douleurs osseuses et parfois fractures)
Les dommages aux cellules des tubules rénaux peuvent être associés à une dégradation des muscles, un ramollissement des os (avec des douleurs osseuses et parfois des fractures), des douleurs musculaires, une faiblesse musculaire et une diminution du potassium ou du phosphate dans le sang.
Si vous pensez ressentir l'un de ces effets secondaires, veuillez contacter votre médecin.
Effets secondaires plus fréquents
Les effets indésirables suivants sont très fréquents (survenant chez au moins 10 patients sur 100) :
- diarrhée, vomissements, nausées, vertiges, maux de tête, éruption cutanée
- sensation de faiblesse, faiblesse musculaire
Les analyses peuvent également montrer :
- réduction des phosphates sanguins
- créatine kinase élevée
Autres effets secondaires possibles
Les effets indésirables suivants sont fréquents (survenant chez un maximum de 10 patients sur 100 patients) :
- douleur, maux d'estomac
- difficulté à dormir, cauchemars
- problèmes digestifs résultant de malaise après les repas, sensation de satiété, gaz intestinaux
- éruptions cutanées (y compris des taches rouges ou des pustules parfois accompagnées de cloques et d'un gonflement de la peau), qui peuvent être une réaction allergique, une sensation de brûlure, un changement de couleur de la peau avec l'apparition de taches sombres.
- autres réactions allergiques, telles que respiration sifflante, ballonnements ou étourdissements
Les analyses peuvent également montrer :
- diminution du nombre de globules blancs (cela peut vous rendre plus vulnérable aux infections)
- augmentation des triglycérides (acides gras), de la bile ou du glucose dans le sang
- problèmes de foie et de pancréas
Les effets indésirables suivants sont peu fréquents (survenant chez au maximum 1 patient sur 100) :
- anémie (faible nombre de globules rouges)
- dégradation des muscles, douleurs musculaires ou faiblesse musculaire, qui peuvent survenir à la suite de lésions des cellules des tubules rénaux
Les analyses peuvent également montrer :
- réduction du potassium dans le sang
- augmentation de la créatinine sanguine
- changements dans l'urine
Les effets indésirables suivants sont rares (survenant chez au maximum 1 patient sur 1 000) :
- maux de dos causés par des problèmes rénaux
Autres effets secondaires possibles
Chez les enfants traités par l'emtricitabine, l'un des composants de Truvada, des cas d'anémie (faible nombre de globules rouges) sont fréquemment survenus et une décoloration de la peau, y compris des taches sombres, est très fréquente. Si la production de globules rouges est réduite, un enfant peut ressentir des symptômes tels que fatigue ou essoufflement.
Truvada peut provoquer des changements dans la forme du corps en modifiant la façon dont la graisse corporelle est distribuée. Vous pouvez perdre de la graisse sur vos jambes, vos bras et votre visage ; une prise de graisse autour de l'abdomen (ventre) et des organes internes ; une augmentation mammaire ou une accumulation de graisse dans la nuque (« bosse de bison ») peuvent survenir. La cause et les effets à long terme de ces changements ne sont pas encore connus.
Truvada peut également provoquer une hyperlipémie (augmentation du taux de graisse dans le sang) et une résistance à l'insuline.Votre médecin vous prescrira des tests pour mesurer ces valeurs.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien, y compris tout effet indésirable éventuel non mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration figurant à l'annexe V. En déclarant les effets indésirables, vous contribuez à fournir plus d'informations sur la sécurité de ce médicament.
Expiration et conservation
Gardez ce médicament hors de la vue et de la portée des enfants.
N'utilisez pas ce médicament après la date de péremption indiquée sur le flacon et la boîte après {EXP}. La date d'expiration fait référence au dernier jour de ce mois.
Conserver dans l'emballage d'origine à l'abri de l'humidité Conserver le flacon bien fermé.
Ne jetez aucun médicament au tout-à-l'égout ou avec les ordures ménagères.Demandez à votre pharmacien comment jeter les médicaments que vous n'utilisez plus.Cela contribuera à protéger l'environnement.
Composition et forme pharmaceutique
Ce que contient Truvada
- Les substances actives sont l'emtricitabine et le ténofovir disoproxil. Chaque comprimé pelliculé de Truvada contient 200 mg d'emtricitabine et 245 mg de ténofovir disoproxil (équivalent à 300 mg de fumarate de ténofovir disoproxil ou 136 mg de ténofovir).
- Les autres composants sont la croscarmellose sodique, le triacétate de glycérol (E1518), l'hypromellose (E464), la laque aluminique carmin d'indigo (E132), le lactose monohydraté, le stéarate de magnésium (E572), la cellulose microcristalline (E460), l'amidon prégélatinisé (sans gluten) et le titane. (E171).
A quoi ressemble Truvada et contenu de l'emballage extérieur
Les comprimés pelliculés Truvada sont bleus, en forme de gélule, portant l'inscription "GILEAD" sur une face et le numéro "701" sur l'autre face. Truvada est fourni en flacons de 30 comprimés. Chaque flacon contient du gel de silice sous forme de un déshydratant, qui doit être conservé dans le flacon pour protéger les comprimés. Le gel de silice est contenu dans un sachet ou un pot séparé et ne doit pas être avalé.
Les présentations suivantes sont disponibles : Emballage extérieur contenant 1 flacon de 30 comprimés pelliculés et 90 (3 flacons de 30) comprimés pelliculés. Toutes les présentations peuvent ne pas être commercialisées.
Notice d'emballage source : AIFA (Agence italienne des médicaments). Contenu publié en janvier 2016. Les informations présentes peuvent ne pas être à jour.
Pour avoir accès à la version la plus récente, il est conseillé d'accéder au site Internet de l'AIFA (Agence Italienne du Médicament). Avis de non-responsabilité et informations utiles.
01.0 DÉNOMINATION DU MÉDICAMENT
TRUVADA 200 MG / 245 MG COMPRIMÉS ENVELOPPÉS DE FILM
02.0 COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient 200 mg d'emtricitabine et 245 mg de ténofovir disoproxil (équivalent à 300 mg de fumarate de ténofovir disoproxil ou 136 mg de ténofovir).
Excipient à effet notoire :
Chaque comprimé contient 96 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
03.0 FORME PHARMACEUTIQUE
Comprimé pelliculé.
Comprimé pelliculé bleu, en forme de capsule, mesurant 19 mm x 8,5 mm, gravé « GILEAD » sur une face et « 701 » sur l'autre face.
04.0 INFORMATIONS CLINIQUES
04.1 Indications thérapeutiques
Truvada est une association à dose fixe d'emtricitabine et de fumarate de ténofovir disoproxil.Il est indiqué en association avec un traitement antirétroviral pour le traitement des adultes infectés par le VIH-1 âgés de 18 ans et plus.
La démonstration du bénéfice de l'association de l'emtricitabine et du fumarate de ténofovir disoproxil dans le traitement antirétroviral repose uniquement sur des études chez des patients non prétraités (voir rubrique 5.1).
04.2 Posologie et mode d'administration
Le traitement doit être initié par un médecin expérimenté dans le domaine de l'infection par le VIH.
Dosage
Adultes: La dose recommandée de Truvada est d'un comprimé, pris par voie orale, une fois par jour. Pour optimiser l'absorption du ténofovir, il est recommandé de prendre Truvada avec de la nourriture.Même un repas léger est suffisant pour améliorer l'absorption du ténofovir des comprimés combinés (voir rubrique 5.2).
Si l'arrêt du traitement par l'un des composants de Truvada est indiqué, ou si la dose doit être ajustée, des formulations distinctes d'emtricitabine et de fumarate de ténofovir disoproxil sont disponibles.Veuillez vous référer au Résumé des Caractéristiques du Produit de ces médicaments.
Si le patient oublie une dose de Truvada dans les 12 heures suivant l'heure habituelle de prise, il doit prendre Truvada dès que possible, avec de la nourriture, et continuer avec le schéma posologique habituel. heures et qu'il est presque l'heure de votre prochaine dose, vous ne devez pas prendre la dose oubliée et continuer simplement avec votre schéma posologique habituel.
Si le patient vomit dans l'heure suivant la prise de Truvada, il doit prendre un autre comprimé. Si le patient vomit plus d'une heure après avoir pris Truvada, il n'a pas besoin de prendre une autre dose.
Populations particulières
Les personnes plus âgées: Il n'y a pas de données disponibles sur lesquelles baser une recommandation posologique pour les patients de plus de 65 ans. Cependant, des ajustements de la dose quotidienne recommandée pour les adultes ne devraient pas être nécessaires, à moins qu'il n'y ait des preuves d'insuffisance rénale.
Insuffisance rénale: L'emtricitabine et le ténofovir sont éliminés par excrétion rénale et l'exposition à l'emtricitabine et au ténofovir augmente chez les patients atteints d'insuffisance rénale.Par conséquent, Truvada ne doit être utilisé chez les patients atteints d'insuffisance rénale que si les bénéfices potentiels du traitement peuvent être considérés comme supérieurs aux risques potentiels. Les patients insuffisants rénaux nécessitent une surveillance étroite de la fonction rénale (voir rubrique 4.4). Des ajustements de l'intervalle posologique sont recommandés chez les patients dont la clairance de la créatinine est comprise entre 30 et 49 ml/min. Ces ajustements posologiques n'ont pas été confirmés dans les essais cliniques et, chez ces patients, la réponse clinique au traitement doit être étroitement surveillée (voir rubriques 4.4 et 5.2). .
Insuffisance rénale légère (clairance de la créatinine comprise entre 50 et 80 ml/min) : Peu de données issues d'essais cliniques soutiennent l'administration de Truvada une fois par jour chez les patients atteints d'insuffisance rénale légère (voir rubrique 4.4).
Insuffisance rénale modérée (clairance de la créatinine comprise entre 30 et 49 ml/min) : L'administration de Truvada toutes les 48 heures est recommandée sur la base d'une modélisation à partir des données pharmacocinétiques d'une dose unique avec l'emtricitabine et le fumarate de ténofovir disoproxil, chez des sujets non infectés par le VIH présentant divers degrés d'insuffisance rénale (voir rubrique 4.4).
Insuffisance rénale sévère (hémodialyse clairance de la créatinine : Truvada n'est pas recommandé chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine
Fonction hépatique altérée : La pharmacocinétique du Truvada et de l'emtricitabine n'a pas été étudiée chez les patients présentant une insuffisance hépatique. La pharmacocinétique du ténofovir a été étudiée chez des patients insuffisants hépatiques pour lesquels aucune modification posologique du fumarate de ténofovir disoproxil n'est requise. Sur la base du métabolisme hépatique minimal et de la voie d'élimination rénale de l'emtricitabine, il est peu probable qu'une modification de la dose de Truvada soit nécessaire chez les patients présentant une insuffisance hépatique (voir rubriques 4.4 et 5.2).
Si le traitement par Truvada est interrompu chez des patients co-infectés par le VIH et le VHB, ces patients doivent être étroitement surveillés afin de détecter toute exacerbation de l'hépatite (voir rubrique 4.4).
Population pédiatrique : La sécurité et l'efficacité de Truvada chez les enfants de moins de 18 ans n'ont pas été établies (voir rubrique 5.2).
Mode d'administration
Les comprimés de Truvada doivent être pris une fois par jour, par voie orale, avec de la nourriture.
Si les patients ont des difficultés à avaler, Truvada peut être dissous dans environ 100 ml d'eau, de jus d'orange ou de jus de raisin et pris immédiatement.
04.3 Contre-indications
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique 6.1.
04.4 Mises en garde spéciales et précautions d'emploi appropriées
Co-administration avec d'autres médicaments
Truvada ne doit pas être co-administré avec d'autres médicaments contenant de l'emtricitabine, du ténofovir disoproxil (sous forme de fumarate) ou d'autres analogues de la cytidine, tels que la lamivudine (voir rubrique 4.5). Truvada ne doit pas être administré en concomitance avec l'adéfovir dipivoxil.
Co-administration de fumarate de ténofovir disoproxil et de didanosine : Ce n'est pas recommandé. L'administration concomitante de fumarate de ténofovir disoproxil et de didanosine a entraîné une augmentation de 40 à 60 % de l'exposition systémique à la didanosine, ce qui peut augmenter le risque d'effets indésirables liés à la didanosine (voir rubrique 4.5. Des pancréatites et des acidoses ont été rarement rapportées. lactiques, parfois fatale. L'administration concomitante de fumarate de ténofovir disoproxil et de didanosine à une dose quotidienne de 400 mg a été associée à une diminution significative du nombre de cellules CD4, probablement en raison d'une « interaction intracellulaire qui augmente les taux de didanosine phosphorylée (active). La réduction de la dose de didanosine co-administrée avec le fumarate de ténofovir disoproxil à 250 mg a été associée à un « taux élevé d'échecs virologiques » dans de nombreuses combinaisons testées.
3 thérapie nucléosidique
Lorsque le fumarate de ténofovir disoproxil a été administré en association avec la lamivudine et l'abacavir, ainsi qu'avec la lamivudine et la didanosine dans le schéma posologique une fois par jour, un « taux élevé d'échecs virologiques et d'apparition précoce de résistance ont été observés ». Il existe une étroite similitude structurelle entre la lamivudine et l'emtricitabine et une similitude dans la pharmacocinétique et la pharmacodynamique de ces deux agents. Par conséquent, les mêmes problèmes pourraient survenir si Truvada est administré avec un troisième analogue nucléosidique.
Infections opportunistes
Les patients recevant Truvada ou tout autre traitement antirétroviral peuvent continuer à développer des infections opportunistes et d'autres complications de l'infection par le VIH, ils doivent donc être étroitement surveillés par des médecins expérimentés dans le traitement des patients atteints de maladies associées au VIH.
Transmission du VIH
Bien qu'il ait été démontré qu'une suppression virale efficace avec un traitement antirétroviral réduisait considérablement le risque de transmission sexuelle, un risque résiduel ne peut être exclu. Des précautions doivent être prises pour empêcher la transmission conformément aux directives nationales.
Insuffisance rénale
L'emtricitabine et le ténofovir sont principalement éliminés par les reins via une combinaison de filtration glomérulaire et de sécrétion tubulaire active. Des cas d'insuffisance rénale, d'insuffisance rénale, d'élévation de la créatinine, d'hypophosphatémie et de tubulopathie proximale (y compris le syndrome de Fanconi) ont été rapportés avec l'utilisation du fumarate de ténofovir disoproxil dans la pratique clinique (voir rubrique 4.8).
La mesure de la clairance de la créatinine est recommandée chez tous les patients avant l'instauration du traitement par Truvada et la fonction rénale (clairance de la créatinine et phosphate sérique) doit être surveillée après deux à quatre semaines de traitement, après trois mois de traitement et ensuite tous les trois à six mois chez patients sans facteurs de risque rénaux. Une surveillance plus fréquente de la fonction rénale est nécessaire chez les patients à risque d'insuffisance rénale.
Patients atteints d'insuffisance rénale (clairance de la créatinine) La sécurité rénale avec Truvada n'a été étudiée que dans une mesure limitée chez les patients atteints d'insuffisance rénale (clairance de la créatinine).
Si le phosphate sérique est la glycémie et le potassium et le glucose dans les urines (voir rubrique 4.8, tubulopathie proximale). L'interruption du traitement par Truvada doit également être envisagée chez les patients présentant une clairance de la créatinine inférieure à 50 ml/min ou présentant une diminution de la phosphatémie à L'utilisation de Truvada doit être évitée en cas d'utilisation concomitante ou récente de médicaments néphrotoxiques (voir rubrique 4.5). Dans le cas où l'utilisation concomitante de Truvada et d'agents néphrotoxiques ne peut être évitée, la fonction rénale doit être surveillée chaque semaine. Après l'instauration de doses multiples ou élevées d'anti-inflammatoires non stéroïdiens (AINS), des cas d'insuffisance rénale aiguë ont été rapportés chez des patients traités par le fumarate de ténofovir disoproxil présentant des facteurs de risque de dysfonctionnement rénal. . , la fonction rénale doit être surveillée de manière adéquate. Un risque plus élevé d'insuffisance rénale a été rapporté chez les patients recevant du fumarate de ténofovir disoproxil en association avec un inhibiteur de protéase potentialisé par le ritonavir ou le cobicistat. Chez ces patients, une surveillance attentive de la fonction rénale est nécessaire (voir rubrique 4.5). Chez les patients présentant des facteurs de risque rénaux, la co-administration de fumarate de ténofovir disoproxil avec un inhibiteur de protéase boosté doit être soigneusement envisagée. Patients porteurs de souches du VIH avec mutations L'utilisation de Truvada doit être évitée chez les patients ayant déjà reçu un traitement antirétroviral et ayant des souches du VIH-1 avec la mutation K65R (voir rubrique 5.1). Effets sur l'os Dans une étude contrôlée menée sur 144 semaines, dans laquelle le fumarate de ténofovir disoproxil a été comparé à la stavudine en association avec la lamivudine et l'éfavirenz chez des patients non prétraités par des antirétroviraux, de légères diminutions de la densité minérale osseuse ont été observées au niveau de la hanche et de la colonne vertébrale. Les diminutions de la densité minérale osseuse de la colonne vertébrale et les changements par rapport à la valeur initiale des biomarqueurs osseux étaient significativement plus importants dans le groupe du fumarate de ténofovir disoproxil à la semaine 144. Les diminutions de la densité minérale osseuse de la hanche étaient significativement plus élevées dans ce groupe jusqu'à 96 semaines. Cependant, il n'y avait pas de risque accru de fractures ou de signes d'anomalies osseuses pertinentes après 144 semaines de traitement. Des anomalies osseuses (conduisant rarement à des fractures) peuvent être associées à une tubulopathie rénale proximale (voir rubrique 4.8). Si des anomalies osseuses sont suspectées, une consultation appropriée doit être recherchée. Patients infectés par le VIH co-infectés par le virus de l'hépatite B ou C Les patients atteints d'hépatite B ou C chronique qui sont traités par un traitement antirétroviral ont un risque accru d'effets indésirables hépatiques graves et potentiellement mortels. Les médecins doivent se référer aux directives thérapeutiques actuelles pour le traitement optimal de l'infection par le VIH chez les patients co-infectés par le virus de l'hépatite B (VHB). En cas de traitement antiviral concomitant de l'hépatite B ou C, veuillez également vous référer au résumé des caractéristiques du produit de ces médicaments. L'innocuité et l'efficacité de Truvada n'ont pas été établies pour le traitement de l'infection chronique par le VHB. L'emtricitabine et le ténofovir, individuellement et en association, se sont révélés actifs contre le VHB dans les études pharmacodynamiques (voir rubrique 5.1). Une expérience clinique limitée suggère que l'emtricitabine et le fumarate de ténofovir disoproxil ont une activité anti-VHB lorsqu'ils sont utilisés ensemble dans un traitement antirétroviral combiné pour contrôler l'infection par le VIH. Chez les patients co-infectés par le VIH et le VHB, l'arrêt du traitement par Truvada peut être associé à des exacerbations aiguës sévères de l'hépatite. Les patients co-infectés par le VIH et le VHB qui ont arrêté l'administration de Truvada doivent être étroitement surveillés, avec suivre clinique et biologique, pendant au moins plusieurs mois après l'arrêt du traitement. Le cas échéant, la reprise du traitement contre l'hépatite B peut être justifiée. Chez les patients atteints d'une maladie hépatique avancée ou d'une cirrhose, l'arrêt du traitement n'est pas recommandé car une exacerbation de l'hépatite post-traitement peut conduire à une décompensation hépatique. Maladie du foie La sécurité et l'efficacité de Truvada n'ont pas été établies chez les patients présentant une insuffisance hépatique significative à l'inclusion. La pharmacocinétique du Truvada et de l'emtricitabine n'a pas été étudiée chez les patients présentant une insuffisance hépatique. La pharmacocinétique du ténofovir a été étudiée chez les patients présentant une insuffisance hépatique et pour aucune modification de dose. est nécessaire Compte tenu du métabolisme hépatique minimal et de la voie d'élimination rénale de l'emtricitabine, il est peu probable qu'une modification de la dose de Truvada soit nécessaire chez les patients présentant une insuffisance hépatique (voir rubrique 5.2). Patients présentant un dysfonctionnement hépatique préexistant, y compris une hépatite chronique active, pendant un traitement antirétroviral combiné (thérapie antirétrovirale combinée, CART) montrent une augmentation de la fréquence des anomalies de la fonction hépatique et doivent être surveillés conformément à la pratique clinique courante. En cas d'aggravation de la maladie hépatique chez de tels patients, l'arrêt ou l'arrêt du traitement doit être envisagé. Lipodystrophie Le CART a été associé à la redistribution de la graisse corporelle (lipodystrophie) chez les patients infectés par le VIH. Les conséquences à long terme de ces événements sont actuellement inconnues. La connaissance du mécanisme est incomplète. Une association entre la lipomatose viscérale et les inhibiteurs de protéase et la lipoatrophie et les inhibiteurs nucléosidiques de la transcriptase inverse a été émise.Un risque accru de lipodystrophie a été associé à la présence de facteurs individuels, tels que l'âge avancé, et de facteurs liés au médicament, tels qu'une durée plus longue de traitement antirétroviral et altérations métaboliques associées. L'examen clinique doit inclure une évaluation des signes physiques de redistribution des graisses. Des mesures des lipides sériques et de la glycémie à jeun doivent être envisagées. Les anomalies du métabolisme lipidique doivent être traitées comme cliniquement appropriées (voir rubrique 4.8). Le ténofovir étant structurellement apparenté aux analogues nucléosidiques, le risque de lipodystrophie ne peut être exclu. Cependant, les données cliniques de 144 semaines de traitement chez des patients non prétraités par antirétroviraux indiquent que le risque de lipodystrophie était plus faible avec le fumarate de ténofovir disoproxil qu'avec la stavudine lorsqu'il était administré avec la lamivudine et l'éfavirenz. Dysfonctionnement mitochondrial Il a été prouvé, soit in vivo cette in vitro, que les analogues nucléosidiques et nucléotidiques provoquent des niveaux variables de dommages mitochondriaux. Des cas de dysfonctionnement mitochondrial ont été rapportés chez des nourrissons séronégatifs exposés au VIH, in utero et/ou après la naissance, aux analogues nucléosidiques. Les principaux effets indésirables rapportés sont des altérations hématologiques (anémie, neutropénie), des altérations métaboliques (hyperlactatémie, hyperlipasémie). Ces événements sont souvent transitoires. Certains changements neurologiques (hypertonie, convulsions, comportement anormal) ont été rapportés comme des épisodes tardifs. On ne sait pas actuellement si les changements neurologiques sont transitoires ou permanents. Pour tout enfant exposé in utero aux analogues nucléosidiques ou nucléotidiques, même s'ils sont séronégatifs, un suivre clinique et de laboratoire et, en cas de signes ou de symptômes pertinents, un examen complet pour détecter un éventuel dysfonctionnement mitochondrial. Ces résultats ne modifient pas les recommandations nationales actuelles concernant l'utilisation du traitement antirétroviral chez les femmes enceintes pour prévenir la transmission verticale du VIH. Syndrome de réactivation immunitaire Chez les patients infectés par le VIH présentant un déficit immunitaire sévère au moment de l'instauration du CART, une réaction inflammatoire à des agents pathogènes opportunistes asymptomatiques ou résiduels peut survenir, provoquant des conditions cliniques graves ou une aggravation des symptômes. En règle générale, de telles réactions ont été observées au cours des premières semaines ou des premiers mois après le début du traitement par CART, par exemple la rétinite à cytomégalovirus, les infections mycobactériennes généralisées et/ou focales et la pneumonie. Pneumocystis jirovecii. Tout symptôme inflammatoire doit être évalué et un traitement instauré si nécessaire. La survenue de maladies auto-immunes (telles que la maladie de Graves) a également été rapportée dans le cadre de la réactivation immunitaire ; cependant, le délai d'apparition enregistré est plus variable et ces événements peuvent survenir plusieurs mois après le début du traitement. Les patients infectés par le VIH et co-infectés par le virus de l'hépatite B peuvent présenter des exacerbations aiguës de l'hépatite associées à un syndrome de réactivation immunitaire après le début du traitement antirétroviral. Ostéonécrose Bien que l'étiologie soit considérée comme multifactorielle (incluant l'utilisation de corticoïdes, la consommation d'alcool, une immunosuppression sévère, un indice de masse corporelle plus élevé), des cas d'ostéonécrose ont été rapportés principalement chez des patients atteints d'une maladie VIH avancée et/ou d'une exposition à long terme au CART. Il est conseillé de consulter un médecin en cas d'inconfort, de douleur et de raideur articulaires ou de difficultés de mouvement. Les personnes plus âgées Truvada n'a pas été étudié chez les patients de plus de 65 ans. L'insuffisance rénale est plus probable chez les personnes âgées, c'est pourquoi le traitement par Truvada chez les personnes âgées doit être entrepris avec prudence. Truvada contient du lactose monohydraté. Par conséquent, les patients présentant des problèmes héréditaires rares d'intolérance au galactose, de déficit en Lapp-lactase ou de malabsorption du glucose-galactose ne doivent pas prendre ce médicament. Comme Truvada contient de l'emtricitabine et du fumarate de ténofovir disoproxil, toute interaction observée avec ces substances actives peut également se produire avec Truvada. Les études d'interaction n'ont été réalisées que chez l'adulte. La pharmacocinétique de régime permanent d'emtricitabine et de ténofovir n'a pas été affecté par l'administration concomitante par rapport aux médicaments seuls administrés individuellement. Éducation in vitro et la pharmacocinétique clinique a montré que le potentiel d'interactions médiées par le CYP450 entre l'emtricitabine et le fumarate de ténofovir disoproxil et d'autres médicaments est faible. Thérapies concomitantes déconseillées En raison de la similitude avec l'emtricitabine, Truvada ne doit pas être administré en même temps que d'autres analogues de la cytidine, tels que la lamivudine (voir rubrique 4.4). En tant qu'association médicamenteuse fixe, Truvada ne doit pas être administré en même temps que d'autres médicaments contenant l'une des substances actives, l'emtricitabine ou le fumarate de ténofovir disoproxil. Truvada ne doit pas être administré en concomitance avec l'adéfovir dipivoxil. Didanosine : La co-administration de Truvada et de didanosine n'est pas recommandée (voir rubrique 4.4 et Tableau 1). Médicaments excrétés par le rein : L'emtricitabine et le ténofovir étant principalement éliminés par les reins, la co-administration de Truvada avec des médicaments qui réduisent la fonction rénale ou qui entrent en compétition pour la sécrétion tubulaire active (par exemple le cidofovir) peut augmenter les concentrations sériques d'emtricitabine, de ténofovir et/ou d'autres médicaments co-administrés. des produits. L'utilisation de Truvada doit être évitée en cas d'utilisation concomitante ou récente de médicaments néphrotoxiques. Certains exemples incluent, sans s'y limiter : les aminosides, l'amphotéricine B, le foscarnet, le ganciclovir, la pentamidine, la vancomycine, le cidofovir ou l'interleukine-2 (voir rubrique 4.4). Autres interactions Les interactions entre les composants de Truvada, les inhibiteurs de protéase et les inhibiteurs nucléosidiques de la transcriptase inverse sont présentées dans le tableau 1 ci-dessous ("l'augmentation est indiquée par" ", la diminution par " ↓ ", aucun changement par " ↔ ", deux fois par jour comme " bid " , une fois par jour sous la forme "qd") Lorsqu'ils sont disponibles, les intervalles de confiance à 90 % sont indiqués entre parenthèses. Tableau 1 : Interactions entre les composants individuels de Truvada et d'autres médicaments Études menées avec d'autres médicaments Emtricitabine : In vitro L'emtricitabine n'a inhibé le métabolisme médié par aucune des isoformes humaines du CYP450 suivantes : 1A2, 2A6, 2B6, 2C9, 2C19, 2D6 et 3A4. L'emtricitabine n'a pas inhibé l'enzyme responsable de la glucuronidation. Il n'y a pas d'interactions pharmacocinétiques cliniquement significatives lorsque l'emtricitabine est co-administrée avec l'indinavir, la zidovudine, la stavudine ou le famciclovir. Fumarate de ténofovir disoproxil : L'administration concomitante de lamivudine, d'indinavir, d'éfavirenz, de nelfinavir ou de saquinavir (stimulé par le ritonavir), de méthadone, de ribavirine, de rifampicine, d'adéfovir dipivoxil ou du contraceptif hormonal norgestimate d'éthinylestradiol avec le fumarate de ténofovir disoproxil n'a produit aucune interaction cliniquement significative avec le fumarate. Truvada: L'administration concomitante de tacrolimus et de Truvada n'a entraîné aucune interaction pharmacocinétique cliniquement significative. Grossesse Un nombre modéré de données chez la femme enceinte (entre 300 et 1 000 grossesses exposées) indique qu'il n'y a pas de malformations ou de toxicité fœtale/néonatale associée à l'emtricitabine et au fumarate de ténofovir disoproxil. Les études animales menées avec l'emtricitabine et le fumarate de ténofovir disoproxil n'ont pas montré de toxicité pour la reproduction (voir rubrique 5.3). Par conséquent, si nécessaire, l'utilisation de Truvada pendant la grossesse peut être envisagée. L'heure du repas Il a été démontré que l'emtricitabine et le ténofovir sont excrétés dans le lait maternel. Les informations sur les effets de l'emtricitabine et du ténofovir sur les nouveau-nés/nourrissons sont insuffisantes. Par conséquent, Truvada ne doit pas être utilisé pendant l'allaitement. En règle générale, il est recommandé que les femmes infectées par le VIH n'allaitent en aucun cas leur enfant afin d'éviter la transmission du virus VIH à l'enfant. La fertilité Il n'y a pas de données sur l'effet du Truvada chez l'homme Les études chez l'animal n'indiquent pas d'effets nocifs de l'emtricitabine ou du ténofovir disoproxil sur la fertilité. Aucune étude sur l'aptitude à conduire des véhicules et à utiliser des machines n'a été réalisée.Cependant, les patients doivent être informés que des étourdissements ont été rapportés pendant le traitement par l'emtricitabine et le fumarate de ténofovir disoproxil. Résumé du profil de sécurité Dans un essai clinique randomisé en ouvert (GS-01-934, voir rubrique 5.1), les réactions les plus fréquemment rapportées, considérées comme possiblement ou probablement liées à l'emtricitabine et/ou au fumarate de ténofovir disoproxil étaient les nausées (12 %) et la diarrhée (7 % ). Dans cette étude, le profil d'innocuité de l'emtricitabine et du fumarate de ténofovir disoproxil s'est avéré cohérent avec celui précédemment observé avec les mêmes agents administrés individuellement avec d'autres antirétroviraux. Chez les patients prenant du fumarate de ténofovir disoproxil, des événements rares, une insuffisance rénale, une insuffisance rénale et une tubulopathie rénale proximale (y compris le syndrome de Fanconi), qui entraînent parfois des modifications osseuses (et rarement des fractures), ont été rapportés. Une surveillance de la fonction rénale est recommandée chez les patients prenant Truvada (voir rubrique 4.4). La lipodystrophie est associée au fumarate de ténofovir disoproxil et à l'emtricitabine (voir rubriques 4.4 et 4.8). L'administration concomitante de fumarate de ténofovir disoproxil et de didanosine n'est pas recommandée car elle peut entraîner une augmentation du risque d'effets indésirables (voir rubrique 4.5).De rares cas de pancréatite et d'acidose lactique, parfois fatales, ont été rapportés (voir rubrique 4.4). Chez les patients co-infectés par le VIH et le VHB, l'arrêt du traitement par Truvada peut être associé à des exacerbations aiguës sévères de l'hépatite (voir rubrique 4.4). Tableau des effets indésirables Les effets indésirables des essais cliniques et de l'expérience post-commercialisation, considérés comme au moins possiblement liés au traitement par les composants de Truvada, sont énumérés ci-dessous dans le tableau 2, ventilés par classe d'organe et de système et par fréquence. ordre décroissant de gravité. Les fréquences sont définies comme : très fréquente (≥ 1/10), fréquente (≥ 1/100, Tableau 2 : Tableau des effets indésirables associés aux composants individuels de Truvada sur la base des études cliniques et de l'expérience post-commercialisation 1 Cet effet indésirable peut survenir à la suite d'une tubulopathie rénale proximale. En l'absence de cette affection, elle n'est pas considérée comme liée au fumarate de ténofovir disoproxil. 2 Chez les patients pédiatriques, une décoloration de la peau (augmentation de la pigmentation) a été fréquemment observée lors du traitement par l'emtricitabine. 3 Cet effet indésirable a été identifié lors de la surveillance post-commercialisation mais n'a pas été observé, pour l'emtricitabine, dans les essais cliniques contrôlés randomisés chez l'adulte ou dans la population pédiatrique VIH ou, pour le fumarate de ténofovir disoproxil, dans les essais cliniques randomisés, contrôlés ou programmés. accès. La fréquence a été évaluée par un calcul statistique basé sur le nombre total de patients exposés à l'emtricitabine lors d'essais contrôlés randomisés (n = 1 563) ou au fumarate de ténofovir disoproxil lors d'essais contrôlés randomisés et de programmes d'accès élargi (n = 7 319). Description de certains effets indésirables Insuffisance rénale: Comme Truvada peut provoquer des lésions rénales, une surveillance de la fonction rénale est recommandée (voir rubriques 4.4 et 4.8). La tubulopathie rénale proximale s'est généralement résolue ou s'est améliorée après l'arrêt du fumarate de ténofovir disoproxil. Chez certains patients, cependant, la diminution de la clairance de la créatinine ne s'est pas complètement résolue malgré l'arrêt du fumarate de ténofovir disoproxil. médicaments), la récupération de la fonction rénale est plus susceptible d'être incomplète malgré l'arrêt du fumarate de ténofovir disoproxil (voir rubrique 4.4). Interactions avec la didanosine : La co-administration de fumarate de ténofovir disoproxil et de didanosine n'est pas recommandée car elle entraîne une augmentation de 40 à 60 % de l'exposition systémique à la didanosine et peut augmenter le risque d'effets indésirables liés à la didanosine (voir rubrique 4.5). Une pancréatite et une acidose lactique, parfois mortelles, ont été rarement rapportées. Lipides, lipodystrophie et altérations métaboliques : Le CART a été associé à des anomalies métaboliques telles qu'une hypertriglycéridémie, une hypercholestérolémie, une résistance à l'insuline, une hyperglycémie et une hyperlactatémie (voir rubrique 4.4). Le CART a été associé à la redistribution de la graisse corporelle (lipodystrophie) chez les patients infectés par le VIH, y compris la perte de graisse sous-cutanée périphérique et faciale, une augmentation de la graisse abdominale et viscérale, une « hypertrophie mammaire et » une accumulation de graisse dorsocervicale (bosse de buffle) (voir rubrique 4.4). Syndrome de réactivation immunitaire : Chez les patients infectés par le VIH présentant un déficit immunitaire sévère au moment de l'initiation du CART, une réaction inflammatoire à des infections opportunistes asymptomatiques ou résiduelles peut survenir.Des troubles auto-immuns (comme la maladie de Basedow) ont également été rapportés ; cependant, le délai d'apparition enregistré est plus variable et ces événements peuvent également survenir plusieurs mois après le début du traitement (voir rubrique 4.4). Ostéonécrose : Des cas d'ostéonécrose ont été rapportés principalement chez des patients présentant des facteurs de risque généralement connus, avec une maladie VIH avancée et/ou une exposition à long terme au CART. La fréquence de ces cas est inconnue (voir rubrique 4.4). Population pédiatrique Des données insuffisantes sont disponibles pour les enfants de moins de 18 ans. Truvada n'est pas recommandé dans cette population de patients (voir rubrique 4.2). Autres populations particulières Les personnes plus âgées: Truvada n'a pas été étudié chez les patients de plus de 65 ans. Les patients âgés sont plus susceptibles d'avoir une fonction rénale réduite, c'est pourquoi Truvada doit être utilisé avec prudence lors du traitement de ces patients (voir rubrique 4.4). Patients atteints d'insuffisance rénale : Le fumarate de ténofovir disoproxil pouvant provoquer une toxicité rénale, une surveillance étroite de la fonction rénale est recommandée chez les patients insuffisants rénaux traités par Truvada (voir rubriques 4.2, 4.4 et 5.2). Les patients co-infecté par le VIH/VHB ou VHC : Dans l'étude GS-01-934, seul un nombre limité de patients étaient co-infectés par le VHB (n = 13) ou le VHC (n = 26). Le profil des effets indésirables de l'emtricitabine et du fumarate de ténofovir disoproxil chez les patients co-infectés par le VIH/VHB ou le VIH/VHC était similaire à celui observé chez les patients infectés par le VIH sans co-infection par le VHB. Cependant, comme prévu dans cette population de patients, des élévations des taux d'AST et d'ALT se sont produites plus fréquemment que dans la population générale infectée par le VIH. Exacerbations d'hépatite après arrêt du traitement : Des preuves cliniques et biologiques d'exacerbations de l'hépatite sont apparues après l'arrêt du traitement chez des patients infectés par le VIH et co-infectés par le VHB (voir rubrique 4.4). Déclaration des effets indésirables suspectés La déclaration des effets indésirables suspectés survenant après l'autorisation du médicament est importante car elle permet un suivi continu du rapport bénéfice/risque du médicament.Les professionnels de santé sont invités à déclarer tout effet indésirable suspecté via le système national de déclaration : Agence italienne des médicaments Site Web : http://www.agenziafarmaco.gov.it/it/responsabili En cas de surdosage, il est nécessaire de surveiller le patient pour détecter tout signe de toxicité (voir rubrique 4.8) et, si nécessaire, d'appliquer les soins de soutien habituels. Jusqu'à 30 % de la dose d'emtricitabine et environ 10 % de la dose de ténofovir peuvent être éliminés par hémodialyse. On ne sait pas si l'emtricitabine peut être éliminée par dialyse péritonéale. Groupe pharmacothérapeutique : Antiviraux à usage systémique ; antiviraux pour le traitement des infections à VIH, combinaisons. Code ATC : J05AR03 Mécanisme d'action et effets pharmacodynamiques L'emtricitabine est un analogue nucléosidique synthétique de la cytidine. Le fumarate de ténofovir disoproxil est converti in vivo dans la substance active ténofovir, qui est un analogue nucléosidique monophosphate (nucléotide) de l'adénosine monophosphate. L'emtricitabine et le ténofovir ont tous deux une activité spécifique contre le virus de l'immunodéficience humaine (VIH-1 et VIH-2) et le virus de l'immunodéficience humaine. l'hépatite B. L'emtricitabine et le ténofovir sont phosphorylés par des enzymes cellulaires pour former respectivement l'emtricitabine triphosphate et le ténofovir diphosphate. Éducation in vitro ont montré que l'emtricitabine et le ténofovir peuvent être entièrement phosphorylés lorsqu'ils sont combinés ensemble dans des cellules. L'emtricitabine triphosphate et le ténofovir diphosphate inhibent de manière compétitive la transcriptase inverse du VIH-1, provoquant une rupture de la chaîne d'ADN. L'emtricitabine triphosphate et le ténofovir diphosphate sont tous deux de faibles inhibiteurs de l'ADN polymérase des mammifères et il n'y a eu aucune preuve de toxicité pour les mitochondries ni in vitro ni in vivo. Activité antivirale in vitro : L'association d'emtricitabine et de ténofovir a été observée in vitro une "activité antivirale synergique.Dans les études d'association avec des inhibiteurs de protéase et avec des analogues nucléosidiques et non nucléosidiques des inhibiteurs de la transcriptase inverse du VIH, des effets synergiques supplémentaires ont été observés. Résistance : In vitro et une résistance a été observée chez certains patients infectés par le VIH-1 en raison du développement de la mutation M184V/I avec l'emtricitabine ou de la mutation K65R avec le ténofovir. Les virus résistants à l'aemtricitabine avec la mutation M184V/I présentaient une résistance croisée à la lamivudine mais conservaient une sensibilité à la didanosine, la stavudine, le ténofovir et la zidovudine. La mutation K65R peut également être sélectionnée par l'abacavir ou la didanosine et entraîner une sensibilité réduite à ces agents plus la lamivudine, l'emtricitabine et le ténofovir. Le fumarate de ténofovir disoproxil doit être évité chez les patients VIH-1 porteurs de la mutation K65R. De plus, une substitution K70E dans la transcriptase inverse du VIH-1 a été sélectionnée avec le ténofovir, entraînant une sensibilité légèrement réduite à l'abacavir, à l'emtricitabine, à la lamivudine et au ténofovir. Les patients infectés par le VIH-1 qui présentent au moins 3 mutations associées à l'analogue de la thymidine (TAM), y compris des mutations de la transcriptase inverse M41L ou L210W, ont présenté une sensibilité réduite au fumarate de ténofovir disoproxil. Résistance in vivo (patients non préalablement traités par antirétroviraux) : Dans un essai clinique randomisé en ouvert (GS-01-934) chez des patients naïfs d'antirétroviraux, un génotypage a été réalisé sur des échantillons de plasma VIH-1 isolés de tous les patients avec un ARN VIH confirmé > 400 copies/ml à 48e, 96e ou 144e semaine ou au moment de l'arrêt prématuré du traitement. A partir de la 144ème semaine : • La mutation M184/I s'est développée dans 2 des 19 (10,5%) souches testées isolées des patients du groupe emtricitabine/ténofovir disoproxil fumarate/éfavirenz et dans 10 des 29 (34,5 %) souches testées isolées du groupe traité lamivudine/zidovudine/éfavirenz (p Fisher Exact
04.5 Interactions avec d'autres médicaments et autres formes d'interactions
Médicament par domaine thérapeutique Effets sur les taux de médicament Variation moyenne en pourcentage de l'ASC, de la Cmax, de la Cmin avec un intervalle de confiance à 90 % si disponible (mécanisme) Recommandation concernant la co-administration avec Truvada (emtricitabine 200 mg, fumarate de ténofovir disoproxil 300 mg) ANTI-INFECTIEUX Antirétroviraux Inhibiteurs de protéase Atazanavir / Ritonavir / Fumarate de ténofovir disoproxil (300 mg q.d./100 mg q.d./300 mg q.d.) Atazanavir : Aucun ajustement posologique n'est recommandé. Une exposition accrue au ténofovir peut potentialiser les événements indésirables associés, y compris des troubles rénaux. La fonction rénale doit être étroitement surveillée (voir rubrique 4.4). ASC : ↓ 25 % (↓ 42 à ↓ 3) Cmax : ↓ 28 % (↓ 50 à ↑ 5) Cmin : ↓ 26 % (↓ 46 à ↑ 10) Ténofovir : ASC : ↑ 37 % Cmax : ↑ 34 % Cmin : ↑ 29 % Atazanavir / Ritonavir / Emtricitabine Interaction non étudiée. Darunavir / Ritonavir / Fumarate de ténofovir disoproxil (300 mg q.d./100 mg q.d./300 mg q.d.) Darunavir : Aucun ajustement posologique n'est recommandé. Une augmentation de l'exposition au ténofovir peut potentialiser les événements indésirables associés, y compris des troubles rénaux. La fonction rénale doit être étroitement surveillée (voir rubrique 4.4). ASC : Cmin : Ténofovir : ASC : ↑ 22% Cmin : ↑ 37 % Darunavir / Ritonavir / Emtricitabine Interaction non étudiée. Lopinavir / Ritonavir / Fumarate de ténofovir disoproxil (400 mg b.i.d./100 mg b.i.d./300 mg q.d.) Lopinavir/Ritonavir : Aucun ajustement posologique n'est recommandé. Une exposition accrue au ténofovir peut potentialiser les événements indésirables associés, y compris des troubles rénaux. La fonction rénale doit être étroitement surveillée (voir rubrique 4.4). ASC : Cmax : Cmin : Ténofovir : ASC : ↑ 32 % (↑ 25 à ↑ 38) Cmax : Cmin : ↑ 51 % (↑ 37 à ↑ 66) Lopinavir / Ritonavir / Emtricitabine Interaction non étudiée. INTI Didanosine / Fumarate de ténofovir disoproxil L'administration concomitante de fumarate de ténofovir disoproxil et de didanosine a entraîné une augmentation de 40 à 60 % de l'exposition systémique à la didanosine, ce qui peut augmenter le risque d'effets indésirables liés à la didanosine. De rares cas de pancréatite et d'acidose lactique, parfois fatales, ont été rapportés. l'administration de fumarate de ténofovir disoproxil et de didanosine à une dose quotidienne de 400 mg a été associée à une diminution significative du nombre de cellules CD4, probablement en raison d'une « interaction intracellulaire qui augmente les niveaux de didanosine phosphorylée (active) ». La réduction de la dose de didanosine co-administrée avec le fumarate de ténofovir disoproxil à 250 mg a été associée à un « taux élevé d'échecs virologiques » dans de nombreuses combinaisons testées pour le traitement de l'infection par le VIH. La co-administration de Truvada et de didanosine n'est pas recommandée (voir rubrique 4.4).
Didanosine / Emtricitabine Interaction non étudiée.
04.6 Grossesse et allaitement
04.7 Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
04.8 Effets indésirables
La fréquence Emtricitabine Fumarate de ténofovir disoproxil Troubles du système sanguin et lymphatique : Commun: neutropénie Rare: anémie 2 Troubles du système immunitaire : Commun: réaction allergique Troubles du métabolisme et de la nutrition : Très commun: hypophosphatémie 1 Commun: hyperglycémie, hypertriglycéridémie Rare: hypokaliémie 1 Rare: acidose lactique Troubles psychiatriques: Commun: insomnie, cauchemars Troubles du système nerveux : Très commun: mal de tête vertiges Commun: vertiges mal de tête Problèmes gastro-intestinaux: Très commun: diarrhée, nausées diarrhée, vomissements, nausées Commun: augmentation de l'amylase, y compris augmentation de l'amylase pancréatique, augmentation de la lipase sérique, vomissements, douleurs abdominales, dyspepsie douleurs abdominales, distension abdominale, flatulences Rare: pancréatite Troubles hépatobiliaires : Commun: augmentation de l'aspartate aminotransférase (AST) sérique et/ou augmentation de l'alanine aminotransférase (ALT) sérique, hyperbilirubinémie augmentation des transaminases Rare: foie gras, hépatite Troubles de la peau et du tissu sous-cutané : Très commun: éruption Commun: éruption vésiculobulleuse, éruption pustuleuse, éruption maculopapuleuse, éruption cutanée, prurit, urticaire, décoloration de la peau (hyperpigmentation) 2 Rare: œdème de Quincke 3 Rare: œdème de Quincke Troubles musculosquelettiques et du tissu conjonctif : Très commun: créatine kinase élevée Rare: rhabdomyolyse1, faiblesse musculaire1 Rare: ostéomalacie (se manifestant par des douleurs osseuses et contribuant rarement aux fractures) 1,3, myopathie1 Troubles rénaux et urinaires : Rare: augmentation de la créatinine, protéinurie Rare: insuffisance rénale (aiguë et chronique), nécrose tubulaire aiguë, tubulopathie rénale proximale y compris syndrome de Fanconi, néphrite (y compris néphrite interstitielle aiguë) 3, diabète insipide néphrogénique Troubles généraux et anomalies au site d'administration : Très commun: asthénie
Commun: douleur, asthénie
04.9 Surdosage
05.0 PROPRIÉTÉS PHARMACOLOGIQUES
05.1 Propriétés pharmacodynamiques
• Aucun virus testé ne contenait la mutation K65R ou K70E.
• Résistance génotypique à l'éfavirenz, principalement la mutation K103N, développée dans le virus de 13 patients sur 19 (68 %) dans le groupe emtricitabine/fumarate de ténofovir disoproxil/éfavirenz et dans le virus de 21 patients sur 29 (72 %) de la comparaison grouper.
Efficacité et sécurité cliniques
Dans un essai clinique randomisé en ouvert (GS-01-934), les patients infectés par le VIH-1 non préalablement traités par antirétroviraux ont été traités soit par un schéma « une fois par jour » composé d'emtricitabine, de fumarate de ténofovir disoproxil et d'éfavirenz (n = 255) ou une association à dose fixe composée de lamivudine et de zidovudine (Combivir) administrées deux fois par jour et d'éfavirenz une fois par jour (n = 254) Les patients du groupe emtricitabine et fumarate de ténofovir disoproxil ont reçu Truvada et éfavirenz Semaine 96 à 144. À l'inclusion, groupes randomisés avaient une médiane plasmatique similaire d'ARN du VIH-1 (5,02 et 5,00 log10 copies / ml) et de nombres de CD4 (233 et 241 cellules / mm3). Le critère principal d'évaluation de l'efficacité de cette étude était l'obtention et le maintien de concentrations validées d'ARN du VIH-1
Comme indiqué dans le tableau 3, les données du critère d'évaluation principal à la semaine 48 ont démontré que l'association d'emtricitabine, de fumarate de ténofovir disoproxil et d'éfavirenz avait une efficacité antivirale supérieure par rapport à l'association à dose fixe de lamivudine et de zidovudine (Combivir) avec l'éfavirenz. Le tableau 3 présente également les données relatives à l'objectif secondaire à la 144e semaine.
Tableau 3 : Données d'efficacité à la semaine 48 et 144 de l'étude GS-01-934 dans laquelle l'emtricitabine, le fumarate de ténofovir disoproxil et l'éfavirenz ont été administrés à des patients infectés par le VIH-1 non préalablement traités par antirétroviraux
* Les patients traités par emtricitabine, fumarate de ténofovir disoproxil et éfavirenz ont reçu Truvada plus éfavirenz de la semaine 96 à la semaine 144.
** La valeur p pour le nombre de cellules CD4 de base est basée sur le test stratifié de Cochran-Mantel-Haenszel
TLOVR = Délai jusqu'à la perte de la réponse virologique
a : Tester Van Elteren
Dans une étude randomisée distincte (M02-418), cent quatre-vingt-dix adultes non prétraités ont été traités une fois par jour par l'emtricitabine et le fumarate de ténofovir disoproxil en association avec le lopinavir/ritonavir administrés une ou deux fois par jour. A 48 semaines, 70% et 64% des patients présentaient un ARN VIH-1 3 et +196 cellules/mm3 respectivement avec les schémas lopinavir/ritonavir, une ou deux fois par jour, respectivement.
L'expérience limitée chez les patients co-infectés par le VIH et le VHB suggère que le traitement par l'emtricitabine ou le fumarate de ténofovir disoproxil en association avec un traitement antirétroviral pour contrôler l'infection par le VIH entraîne également une réduction de l'ADN du VHB (réductions de 3 log10 ou de 4 à 5 log10, respectivement) (voir rubrique 4.4).
Population pédiatrique
La sécurité et l'efficacité de Truvada chez les enfants de moins de 18 ans n'ont pas été établies.
05.2 "Propriétés pharmacocinétiques
Absorption
La bioéquivalence d'un comprimé pelliculé de Truvada avec une gélule d'emtricitabine 200 mg et un comprimé pelliculé de fumarate de ténofovir disoproxil 245 mg a été évaluée après administration d'une dose unique chez des sujets sains à jeun. Après administration orale de Truvada à des sujets sains, l'emtricitabine et le fumarate de ténofovir disoproxil sont rapidement absorbés et le fumarate de ténofovir disoproxil est converti en ténofovir. Des concentrations maximales d'emtricitabine et de ténofovir ont été observées dans le sérum dans les 0,5 à 3,0 heures suivant l'administration à jeun. L'administration de Truvada avec de la nourriture entraîne un retard d'environ trois quarts d'heure pour atteindre la concentration maximale de ténofovir et une augmentation du ténofovir. Une ASC et une Cmax d'environ 35 % et 15 %, respectivement, lorsqu'elles sont administrées avec un repas riche en graisses ou léger, par rapport à une administration à jeun. Pour optimiser l'absorption du ténofovir, il est recommandé de prendre Truvada avec de la nourriture.
Distribution
Après administration intraveineuse, le volume de distribution de l'emtricitabine et du ténofovir a été estimé à environ 1,4 L/kg et 800 ml/kg, respectivement. Après administration orale d'emtricitabine et de fumarate de ténofovir disoproxil, l'emtricitabine et le ténofovir sont largement distribués dans l'organisme. In vitro La liaison in vitro de l'emtricitabine aux protéines plasmatiques humaines était inférieure à 0,7 et 7,2 % des protéines du ténofovir aux protéines plasmatiques ou sériques, respectivement.
Biotransformation
Le métabolisme de l'emtricitabine est limité. La biotransformation de l'emtricitabine comprend l'oxydation du groupe thiol pour former des diastéréomères 3"-sulfoxyde (environ 9 % de la dose) et la conjugaison avec l'acide glucuronique pour former du 2" -O-glucuronide (environ 4 % de la dose). Études in vitro ont déterminé que ni le fumarate de ténofovir disoproxil ni le ténofovir ne sont des substrats des enzymes du CYP450. Ni l'emtricitabine ni le ténofovir n'inhibent in vitro métabolisme des médicaments médié par l'une des principales isoformes humaines du CYP450 impliquées dans la biotransformation des médicaments. De plus, l'emtricitabine n'inhibe pas l'uridine-5"-diphosphoglucuronyltransférase, l'enzyme responsable de la glucuronidation.
Élimination
L'emtricitabine est principalement excrétée par les reins, avec récupération complète de la dose obtenue dans les urines (environ 86 %) et les selles (environ 14 %). Treize pour cent de la dose d'emtricitabine sont récupérés dans l'urine sous forme de trois métabolites. La clairance systémique de l'emtricitabine est en moyenne de 307 ml/min. Après administration orale, la demi-vie d'élimination de l'emtricitabine est d'environ 10 heures.
Le ténofovir est éliminé principalement par les reins par filtration et par un système de transport tubulaire actif, environ 70 à 80 % de la dose étant excrétée sous forme inchangée dans les urines après administration intraveineuse. La clairance apparente du ténofovir était d'environ 307 ml/min. La clairance rénale a été estimée. être d'environ 210 mL/min, ce qui est supérieur au débit de filtration glomérulaire, indiquant que la sécrétion tubulaire active est un élément important dans l'élimination du ténofovir. Après administration orale, la demi-vie d'élimination du ténofovir était d'environ 12 à 18 heures.
Les personnes plus âgées
Aucune étude pharmacocinétique avec l'emtricitabine et le ténofovir n'a été menée chez les personnes âgées (plus de 65 ans).
Sexe
La pharmacocinétique de l'emtricitabine et du ténofovir est similaire chez les hommes et les femmes.
Ethnicité
Aucune différence pharmacocinétique cliniquement significative liée à l'origine ethnique n'a été identifiée pour l'emtricitabine.La pharmacocinétique du ténofovir à travers les groupes ethniques n'a pas été spécifiquement étudiée.
Population pédiatrique
En général, la pharmacocinétique de l'emtricitabine chez les nourrissons, les enfants et les adolescents (âgés de 4 mois à 18 ans) est similaire à celle observée chez les adultes. Aucune étude pharmacocinétique n'a été menée avec le ténofovir chez les enfants et les adolescents (âgés de moins de 18 ans).
Insuffisance rénale
Peu de données pharmacocinétiques sont disponibles pour l'emtricitabine et le ténofovir après co-administration dans des formulations séparées ou sous forme de Truvada chez les patients insuffisants rénaux. Les paramètres pharmacocinétiques ont été principalement déterminés après l'administration d'une dose unique d'emtricitabine à 200 mg ou de ténofovir disoproxil à 245 mg à des patients non infectés par le VIH présentant divers degrés d'insuffisance rénale. Le degré d'insuffisance rénale était défini par la clairance de la créatinine (CrCl) (fonction rénale normale lorsque CrCl > 80 mL/min ; insuffisance légère avec CrCl = 50-79 mL/min ; insuffisance modérée avec CrCl = 30-49 mL/min). min et atteinte sévère avec CrCl = 10-29 mL/min).
L'exposition moyenne (% CV) à l'emtricitabine est passée de 12 (25 %) mcg•h/ml chez les sujets ayant une fonction rénale normale à 20 (6 %) mcg•h/ml, 25 (23 %) mcg•h/ml et 34 (6%) mcg • h / ml, respectivement, chez les patients atteints d'insuffisance rénale légère, modérée et sévère.
L'exposition moyenne (% CV) au ténofovir est passée de 2 185 (12 %) ng•h/mL chez les patients ayant une fonction rénale normale à 3 064 (30 %) ng•h/mL, 6 009 (42 %) ng•h/ml et 15 985 ( 45%) ng • h / ml chez les patients atteints d'insuffisance rénale légère, modérée et sévère, respectivement.
L'augmentation de l'éventail des doses de Truvada chez les patients présentant une insuffisance rénale modérée devrait produire des concentrations plasmatiques maximales plus élevées et une Cmin plus faible que chez les patients ayant une fonction rénale normale.
Chez les patients atteints d'insuffisance rénale terminale (IRT) nécessitant une hémodialyse, l'exposition médicamenteuse entre les dialyses augmente substantiellement à 53 (19 %) mcg•h/ml sur 72 heures pour l'emtricitabine, et à 42 857 (29 %) ng•h/ml de ténofovir sur 48 heures.
La modification de l'intervalle entre les doses de Truvada est recommandée chez les patients dont la clairance de la créatinine est comprise entre 30 et 49 ml/min. Truvada n'est pas approprié pour les patients présentant une ClCr
Une petite étude clinique a été menée pour évaluer l'innocuité, l'activité antivirale et la pharmacocinétique du fumarate de ténofovir disoproxil en association avec l'emtricitabine chez des patients infectés par le VIH présentant une insuffisance rénale. Un sous-groupe de patients avec une clairance initiale de la créatinine comprise entre 50 et 60 ml/min sous traitement une fois par jour présentait une exposition au ténofovir 2 à 4 fois plus élevée et une aggravation de la fonction rénale.
Insuffisance hépatique
La pharmacocinétique de Truvada n'a pas été étudiée chez les patients présentant une insuffisance hépatique. Cependant, il est peu probable qu'un ajustement de la dose de Truvada soit nécessaire chez les patients présentant une insuffisance hépatique.
La pharmacocinétique de l'emtricitabine n'a pas été étudiée chez des sujets non infectés par le VHB présentant divers degrés d'insuffisance hépatique. En général, la pharmacocinétique de l'emtricitabine chez les sujets infectés par le VHB était similaire à celle des sujets sains et infectés par le VIH.
Une dose unique de 245 mg de ténofovir disoproxil a été administrée à des patients non infectés par le VIH présentant divers degrés d'insuffisance hépatique tels que définis par la classification de Child-Pugh-Turcotte (CPT). La pharmacocinétique du ténofovir n'a pas été sensiblement modifiée chez les sujets atteints d'insuffisance hépatique, ce qui suggère qu'aucun ajustement posologique n'est nécessaire chez ces sujets. Les valeurs moyennes (% CV) de la Cmax et de l'ASC0-∞ du ténofovir étaient de 223 (34,8%) ng/mL et 2 050 (50,8%) ng•h/mL chez les sujets normaux, respectivement, contre 289 (46,0%) ng/ mL et 2 310 (43,5 %) ng•h/mL chez les sujets atteints d'insuffisance hépatique modérée et 305 (24,8 %) ng/mL et 2 740 (44,0 %) ng•h/ml chez les sujets atteints d'insuffisance hépatique sévère.
05.3 Données de sécurité précliniques
Emtricitabine : les données précliniques sur l'emtricitabine ne révèlent aucun risque particulier pour l'homme sur la base des études conventionnelles de pharmacologie de sécurité, toxicité à doses répétées, génotoxicité, potentiel cancérigène et toxicité pour la reproduction et le développement.
Fumarate de ténofovir disoproxil: études précliniques de pharmacologie de sécurité sur le fumarate de ténofovir disoproxil n'ont révélé aucun risque particulier pour l'homme. concentration en phosphates. La toxicité osseuse a été diagnostiquée comme une ostéomalacie (chez le singe) et une diminution de la densité minérale osseuse (densité minérale osseuse, DMO) (chez le rat et le chien). Chez les rats et les jeunes chiens adultes, une toxicité osseuse s'est produite à des expositions ≥ 5 fois l'exposition des patients pédiatriques ou adultes ; chez les jeunes singes infectés, une toxicité osseuse s'est produite à des expositions très élevées après administration sous-cutanée (≥ 40 fois l "exposition du patient). Les résultats d'études chez le rat et le singe suggèrent une réduction liée à la substance de l'absorption intestinale du phosphate, avec une réduction secondaire potentielle de la DMO.
Les études de génotoxicité ont donné des résultats de test positifs in vitro sur le lymphome de souris résultats équivoques dans l'une des souches utilisées dans le test d'Ames et résultats faiblement positifs dans un test USD dans des hépatocytes primaires de rat. Cependant, il était négatif dans l'induction de mutations dans un test du micronoyau de moelle osseuse de souris. in vivo.
Des études de cancérogénicité par voie orale chez le rat et la souris ont montré une faible incidence de tumeurs duodénales à une dose extrêmement élevée chez la souris. Il est peu probable que ces tumeurs soient pertinentes pour l'homme.
Les études de toxicité pour la reproduction réalisées chez le rat et le lapin n'ont révélé aucun effet sur l'accouplement, la fertilité, la gestation ou les paramètres fœtaux. Cependant, dans les études de toxicité périnatale et postnatale, le fumarate de ténofovir disoproxil a réduit la viabilité et le poids des petits aux doses toxiques pour la mère.
Association d'emtricitabine et de fumarate de ténofovir disoproxil : aucune exacerbation des effets toxicologiques n'a été observée dans les études de génotoxicité et les études de toxicité à doses répétées d'une durée allant jusqu'à un mois sur la combinaison de ces deux composants par rapport aux études menées avec les composants individuels.
06.0 INFORMATIONS PHARMACEUTIQUES
06.1 Excipients
Noyau de la tablette :
Croscarmellose sodique
Lactose monohydraté
Stéarate de magnésium (E572)
Cellulose microcristalline (E460)
Amidon prégélatinisé (sans gluten)
Film de revêtement :
Triacétate de glycérol (E1518)
Hypromellose (E464)
Laque d'aluminium carmin d'indigo (E132)
Lactose monohydraté
Dioxyde de titane (E171)
06.2 Incompatibilité
Non pertinent.
06.3 Durée de validité
4 années.
06.4 Précautions particulières de conservation
Conserver dans l'emballage d'origine à l'abri de l'humidité Conserver le flacon bien fermé.
06.5 Nature du conditionnement primaire et contenu de l'emballage
Flacon en polyéthylène haute densité (PEHD) avec un bouchon sécurité enfant en polypropylène contenant 30 comprimés pelliculés et avec un gel de silice comme déshydratant.
Les présentations suivantes sont disponibles : emballage extérieur contenant 1 flacon de 30 comprimés pelliculés et emballage extérieur contenant 90 (3 flacons de 30) comprimés pelliculés. Toutes les présentations peuvent ne pas être commercialisées.
06.6 Instructions d'utilisation et de manipulation
Les médicaments non utilisés et les déchets dérivés de ce médicament doivent être éliminés conformément aux réglementations locales.
07.0 TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHE
Gilead Sciences International Limited
Cambridge
CB21 6GT
Royaume-Uni
08.0 NUMÉRO D'AUTORISATION DE MISE SUR LE MARCHÉ
UE / 1/04/305/001
UE/1/04/305/002
036716013
09.0 DATE DE PREMIÈRE AUTORISATION OU DE RENOUVELLEMENT DE L'AUTORISATION
Date de première autorisation : 21/02/2005
Date du dernier renouvellement : 20/01/2010
10.0 DATE DE RÉVISION DU TEXTE
05/2015