Ingrédients actifs : Ivabradine
Procoralan 5 mg comprimés pelliculés
Procoralan 7,5 mg comprimés pelliculés
Pourquoi Procoralan est-il utilisé ? Pourquoi est-ce?
Procoralan (ivabradine) est un médicament pour le cœur utilisé pour traiter :
- d'angine de poitrine stable symptomatique (une maladie provoquant des douleurs thoraciques) chez les patients adultes dont la fréquence cardiaque est supérieure ou égale à 70 battements par minute. Il est utilisé chez les patients adultes qui ne tolèrent pas ou ne peuvent pas prendre de médicaments pour le cœur appelés bêta-bloquants. Il est également utilisé en association avec des bêta-bloquants chez les patients adultes dont l'état n'est pas entièrement contrôlé par un bêta-bloquant
- d'insuffisance cardiaque chronique chez les patients adultes dont la fréquence cardiaque est supérieure ou égale à 75 battements par minute.Il est utilisé en association avec un traitement conventionnel, qui comprend un traitement par un bêta-bloquant ou lorsque les bêta-bloquants sont contre-indiqués ou non tolérés.
À propos de « l'angine de poitrine stable » (communément appelée « angine de poitrine »):
L'angine stable est une maladie cardiaque qui survient lorsque le cœur ne reçoit pas assez d'oxygène. Elle apparaît généralement entre 40 et 50 ans. Le symptôme le plus courant de l'angine de poitrine est une douleur ou une gêne thoracique. L'angine est plus susceptible de se produire lorsque le cœur bat rapidement dans des situations telles que «l'activité physique», les émotions, l'exposition au froid ou après avoir mangé. Cette augmentation du rythme cardiaque peut provoquer des douleurs thoraciques chez les personnes souffrant d'angine de poitrine.
Informations sur l'insuffisance cardiaque chronique :
L'insuffisance cardiaque chronique est une maladie cardiaque qui survient lorsque le cœur ne peut pas pomper suffisamment de sang vers le reste du corps.Les symptômes les plus courants de l'insuffisance cardiaque sont l'essoufflement, la fatigue, la fatigue et l'enflure des chevilles.
Comment fonctionne Procoralan ?
Procoralan agit principalement en réduisant la fréquence cardiaque de quelques battements par minute. Cela réduit le besoin du cœur en oxygène, en particulier dans les situations où une crise d'angine de poitrine est plus probable. De cette façon, Procoralan aide à contrôler et à diminuer le nombre d'attaques d'angine de poitrine.
De plus, étant donné qu'une fréquence cardiaque élevée affecte négativement la fonction cardiaque et l'espérance de vie des patients atteints d'insuffisance cardiaque chronique, l'action spécifique d'abaissement de la fréquence cardiaque de l'ivabradine contribue à améliorer la fonction cardiaque et l'espérance de vie chez ces patients.
Contre-indications Quand Procoralan ne doit pas être utilisé
Ne prenez pas Procoralan
- si vous êtes allergique à l'ivabradine ou à l'un des autres composants contenus dans ce médicament
- si votre fréquence cardiaque au repos avant le traitement est trop faible (moins de 70 battements par minute) ;
- si vous souffrez d'un choc cardiogénique (une maladie cardiaque traitée à l'hôpital) ;
- si vous souffrez d'un trouble du rythme cardiaque ;
- si vous avez une crise cardiaque ;
- si vous avez une pression artérielle très basse ;
- si vous souffrez d'angine instable (une forme sévère dans laquelle des douleurs thoraciques surviennent très fréquemment et avec ou sans effort) ;
- si vous souffrez d'une insuffisance cardiaque qui s'est récemment aggravée ;
- si le rythme cardiaque est imposé exclusivement par le stimulateur cardiaque ;
- si vous avez de graves problèmes de foie ;
- si vous prenez déjà des médicaments pour traiter les infections fongiques (tels que le kétoconazole, l'itraconazole), des antibiotiques macrolides (tels que l'iosamycine, la clarithromycine, la télithromycine ou l'érythromycine administrés par voie orale) ou des médicaments pour traiter les infections à VIH (tels que le nelfinavir, le ritonavir) ou la néfazodone ( médicament pour traiter la dépression) ou diltiazem, vérapamil (utilisé pour l'hypertension artérielle ou l'angine de poitrine);
- si vous êtes une femme capable d'avoir des enfants et n'utilisez pas de « contraception appropriée » ;
- si vous êtes enceinte ou essayez d'avoir un enfant ;
- si vous allaitez.
Précautions d'emploi Quelles sont les informations à connaître avant de prendre Procoralan
Adressez-vous à votre médecin ou pharmacien avant de prendre Procoralan.
- si vous souffrez de troubles du rythme cardiaque (tels qu'un rythme cardiaque irrégulier, des palpitations, une augmentation des douleurs thoraciques) ou une fibrillation auriculaire sévère (une forme d'arythmie qui rend le rythme cardiaque irrégulier), ou un « trouble de l'électrocardiogramme (ECG) appelé" syndrome du long QT ",
- si vous vous fatiguez facilement, si vous vous sentez étourdi ou essoufflé (cela peut signifier que votre cœur bat trop lentement),
- si vous souffrez de symptômes de fibrillation auriculaire (anormalement élevés (plus de 110 battements par minute) ou de rythme cardiaque irrégulier au repos sans raison apparente rendant la mesure difficile),
- si vous avez eu un accident vasculaire cérébral récent (attaque cérébrale),
- si vous souffrez d'hypotension légère à modérée,
- si vous souffrez d'une tension artérielle non contrôlée, notamment suite à un changement de traitement antihypertenseur,
- si vous souffrez d'une insuffisance cardiaque sévère ou d'une insuffisance cardiaque avec une anomalie de l'électrocardiogramme (ECG) appelée « bloc de branche »,
- si vous souffrez d'une maladie rétinienne chronique,
- si vous avez des problèmes hépatiques modérés, - si vous avez des problèmes rénaux sévères.
Si l'un des cas ci-dessus vous concerne, parlez-en immédiatement à votre médecin avant ou pendant la prise de Procoralan.
Enfants
Procoralan ne doit pas être utilisé chez les enfants et les adolescents de moins de 18 ans.
Interactions Quels médicaments ou aliments peuvent modifier l'effet de Procoralan
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Assurez-vous d'informer votre médecin si vous prenez l'un des médicaments suivants, car votre dose de procoralan peut devoir être surveillée ou ajustée :
- fluconazole (un médicament antifongique)
- rifampicine (un antibiotique)
- barbituriques (pour l'insomnie ou l'épilepsie)
- phénytoïne (pour l'épilepsie)
- Hypericum perforatum ou millepertuis (produit à base de plantes utilisé pour la dépression)
- médicaments qui allongent l'intervalle QT pour traiter les troubles du rythme ou d'autres affections telles que :
- quinidine, disopyramide, ibutilide, sotalol, amiodarone (pour traiter les troubles du rythme cardiaque)
- bepridil (pour traiter l'angine de poitrine)
- certains types de médicaments pour traiter l'anxiété, la schizophrénie ou d'autres psychoses (tels que le pimozide, la ziprasidone, le sertindole)
- médicaments contre le paludisme (tels que la méfloquine ou l'halofantrine)
- érythromycine intraveineuse (un antibiotique)
- pentamidine (un pesticide)
- cisapride (utilisé pour le reflux gastro-œsophagien)
- Certains types de diurétiques pouvant entraîner une baisse des taux de potassium dans le sang, tels que le furosémide, l'hydrochlorothiazide, l'indapamide (utilisés pour traiter l'œdème, l'hypertension artérielle)
Procoralan avec de la nourriture et des boissons
Évitez le jus de pamplemousse pendant que vous prenez Procoralan.
Avertissements Il est important de savoir que :
La grossesse et l'allaitement
Ne prenez pas Procoralan si vous êtes enceinte ou prévoyez le devenir (voir « Ne prenez jamais Procoralan »). Si vous êtes enceinte et avez pris Procoralan, parlez-en à votre médecin.
Ne prenez pas Procoralan si vous êtes en mesure d'avoir des enfants, sauf si vous utilisez des mesures contraceptives appropriées (voir « Ne prenez jamais Procoralan »).
Ne prenez pas Procoralan si vous allaitez (voir « Ne prenez jamais Procoralan »). Informez votre médecin si vous allaitez ou avez l'intention d'allaiter car l'allaitement doit être interrompu si vous prenez Procoralan.
Si vous êtes enceinte ou si vous allaitez, si vous pensez être enceinte ou prévoyez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant de prendre ce médicament.
Conduire et utiliser des machines
Procoralan peut provoquer des phénomènes visuels lumineux temporaires (une luminosité temporaire dans le champ visuel, voir "Effets secondaires possibles"). Si cela vous arrive, soyez très prudent lorsque vous conduisez ou utilisez des machines, en particulier lorsqu'il peut y avoir des changements soudains d'intensité lumineuse, en particulier lors de la conduite de nuit.
Procoralan contient du lactose
Si votre médecin vous a dit que vous aviez une intolérance à certains sucres, contactez votre médecin avant de prendre ce médicament.
Dose, mode et heure d'administration Comment utiliser Procoralan : Posologie
Prenez toujours ce médicament en suivant exactement les indications de votre médecin ou pharmacien.
En cas de doute, consultez votre médecin ou votre pharmacien. Procoralan doit être pris avec les repas.
Si vous êtes traité pour une angine de poitrine stable
La dose initiale ne doit pas dépasser un comprimé de Procoralan 5 mg deux fois par jour. Si vous présentez toujours des symptômes d'angine de poitrine et que vous tolérez bien la dose quotidienne de 5 mg deux fois par jour, la dose peut être augmentée. La dose d'entretien ne doit pas dépasser 7,5 mg deux fois par jour. Votre médecin vous prescrira la dose. La dose habituelle est de 1 comprimé le matin et un comprimé le soir.Dans certains cas (par exemple si vous êtes âgé), votre médecin peut vous prescrire la moitié de la dose, par exemple un demi-comprimé de 5 mg de Procoralan 5 mg (ce qui correspond à 2,5 mg d'ivabradine) dans le matin et un demi-comprimé à 5 mg le soir.
Si vous êtes traité pour une insuffisance cardiaque chronique
La dose initiale habituelle recommandée est d'un comprimé de Procoralan à 5 mg deux fois par jour, à augmenter si nécessaire à un comprimé de Procoralan à 7,5 mg deux fois par jour. Votre médecin décidera de la dose la plus appropriée. La dose habituelle est d'un comprimé le matin et d'un comprimé le soir. In alcuni casi (ad esempio, se è anziano), il medico potrà prescrivere di dimezzare la dose, ovvero mezza compressa da 5 mg di Procoralan 5 mg (che corrisponde a 2,5 mg di ivabradina) la mattina e mezza compressa da 5 mg le soir.
Surdosage Que faire si vous avez pris trop de Procoralan
Si vous avez pris plus de Procoralan que vous n'auriez dû
Une dose élevée de Procoralan peut vous essouffler ou vous fatiguer car votre rythme cardiaque a trop ralenti. Si cela se produit, contactez immédiatement votre médecin.
Si vous oubliez de prendre Procoralan
Si vous oubliez de prendre une dose de Procoralan, prenez la dose suivante à l'heure habituelle. Ne prenez pas de dose double pour compenser une dose oubliée. Le calendrier imprimé sur la plaquette contenant les comprimés vous aidera à vous rappeler quand vous avez pris votre dose. dernier comprimé par Procoralan.
Si vous arrêtez de prendre Procoralan
Le traitement de l'angine de poitrine ou de l'insuffisance cardiaque chronique étant généralement à vie, vous devez en parler à votre médecin avant d'arrêter de prendre ce médicament. Si vous avez l'impression que l'effet de Procoralan est trop fort ou trop faible, consultez votre médecin ou votre pharmacien. Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou votre pharmacien.
Effets secondaires Quels sont les effets secondaires de Procoralan
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, bien que tout le monde n'y soit pas sujet.
La fréquence des effets indésirables possibles énumérés ci-dessous est décrite selon la convention suivante :
très fréquent : peut affecter plus de 1 patient sur 10
fréquent : peut affecter jusqu'à 1 personne sur 10
peu fréquent : peut affecter jusqu'à 1 patient sur 100
rare : peut affecter jusqu'à 1 patient sur 1 000
très rare : peut affecter jusqu'à 1 patient sur 10 000
fréquence indéterminée : la fréquence ne peut pas être estimée à partir des données disponibles.
Les effets indésirables les plus fréquents survenant avec ce médicament dépendent de la dose et sont liés à son mécanisme d'action :
Très commun:
Phénomènes visuels lumineux (brefs moments d'augmentation de la luminosité, le plus souvent causés par des changements soudains d'intensité lumineuse). Ils peuvent également être décrits comme un halo, des flashs colorés, une dégradation de l'image ou des images multiples. Ces phénomènes se développent généralement au cours des deux premiers mois de traitement, après quoi ils peuvent survenir à plusieurs reprises et disparaître pendant ou après le traitement. Fréquent : Modification de la fonction cardiaque (les symptômes sont un ralentissement de la fréquence cardiaque). Ces phénomènes surviennent surtout dans les 2-3 premiers mois suivant le début du traitement.D'autres effets secondaires ont également été rapportés :
Commun:
Contraction rapide et irrégulière du cœur, perception anormale du rythme cardiaque, tension artérielle non contrôlée, maux de tête, vertiges et vision floue (vision floue).
Rare:
Palpitations et battements cardiaques irréguliers, nausées, constipation, diarrhée, douleurs abdominales, étourdissements (vertiges), difficultés respiratoires (dyspnée), crampes musculaires, modifications des paramètres de laboratoire : taux sanguins élevés d'acide urique, excès d'éosinophiles (un type de globules blancs) et élévation de la créatinine (produit de la dégradation musculaire) dans le sang, éruption cutanée, œdème de Quincke (tel que gonflement du visage, de la langue ou de la gorge, difficulté à respirer ou à avaler), hypotension artérielle, évanouissement, sensation de fatigue, sensation de faiblesse , trace cardiaque anormale à l'ECG, vision double, vision altérée.
Rare:
Urticaire, démangeaisons, rougeur de la peau, malaise.
Très rare:
Rythme cardiaque irrégulier.
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien, y compris tout effet indésirable éventuel non mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration figurant à l'annexe V.* En déclarant les effets indésirables, vous contribuez à fournir plus d'informations sur la sécurité de ce médicament.
Expiration et conservation
Gardez ce médicament hors de la vue et de la portée des enfants.
N'utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la plaquette après « EXP ». La date de péremption fait référence au dernier jour du mois.
Ce médicament ne nécessite aucune condition particulière de conservation.
Ne jetez aucun médicament au tout-à-l'égout ou avec les ordures ménagères.Demandez à votre pharmacien comment jeter les médicaments que vous n'utilisez plus.Cela contribuera à protéger l'environnement.
Ce que contient Procoralan
- La substance active est l'ivabradine (sous forme de chlorhydrate).Procoralan 5 mg : Un comprimé pelliculé contient 5 mg d'ivabradine (équivalent à 5,390 mg de chlorhydrate d'ivabradine). Procoralan 7,5 mg : Un comprimé pelliculé contient 7,5 mg d'ivabradine (équivalent à 8,085 mg de chlorhydrate d'ivabradine).
- Les autres composants du noyau du comprimé sont : lactose monohydraté, stéarate de magnésium (E470B), amidon de maïs, maltodextrine, silice colloïdale anhydre (E551), et dans l'enrobage du comprimé : hypromellose (E464), dioxyde de titane (E171), macrogol 6000 , glycérol (E422), stéarate de magnésium (E470B), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Qu'est-ce que Procoralan et contenu de l'emballage extérieur
Les comprimés de Procoralan 5 mg sont de couleur saumon, oblongs, pelliculés, sécables des deux côtés, gravés "5" d'un côté et de l'autre.
Les comprimés de Procoralan 7,5 mg sont des comprimés pelliculés triangulaires de couleur saumon portant l'inscription « 7,5 » gravée sur une face et sur l'autre.
Les comprimés sont disponibles en boîtes calendrier (plaquettes thermoformées en aluminium/PVC) contenant 14, 28, 56, 84, 98, 100 ou 112 comprimés. Toutes les présentations peuvent ne pas être commercialisées.
Notice d'emballage source : AIFA (Agence italienne des médicaments). Contenu publié en janvier 2016. Les informations présentes peuvent ne pas être à jour.
Pour avoir accès à la version la plus à jour, il est conseillé d'accéder au site Internet de l'AIFA (Agence Italienne du Médicament). Avis de non-responsabilité et informations utiles.
01.0 DÉNOMINATION DU MÉDICAMENT
PROCORALAN 5 MG COMPRIMÉS ENROBÉS DE FILM
▼ Médicament faisant l'objet d'une surveillance supplémentaire. Cela permettra l'identification rapide de nouvelles informations de sécurité. Les professionnels de santé sont priés de signaler tout effet indésirable suspecté. Voir rubrique 4.8 pour plus d'informations sur la manière de déclarer les effets indésirables.
02.0 COMPOSITION QUALITATIVE ET QUANTITATIVE
Un comprimé pelliculé contient 5 mg d'ivabradine (équivalent à 5,390 mg d'ivabradine sous forme de chlorhydrate).
Excipient à effet notoire: 63,91 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
03.0 FORME PHARMACEUTIQUE
Comprimé pelliculé.
Comprimé pelliculé de couleur saumon, oblong, sécable sur les deux faces, gravé "5" sur une face.
Le comprimé peut être divisé en moitiés égales.
04.0 INFORMATIONS CLINIQUES
04.1 Indications thérapeutiques
Traitement symptomatique de l'angine de poitrine stable chronique.
L'ivabradine est indiquée dans le traitement symptomatique de l'angine de poitrine chronique stable chez l'adulte présentant une maladie coronarienne, un rythme sinusal normal et une fréquence cardiaque ≥ 70 bpm. L'ivabradine est indiquée :
- chez l'adulte intolérant ou présentant une contre-indication à l'utilisation des bêtabloquants
- soit en association avec des bêta-bloquants chez les patients insuffisamment contrôlés par une dose optimale de bêta-bloquant
Traitement de l'insuffisance cardiaque chronique
L'ivabradine est indiquée dans l'insuffisance cardiaque chronique classe NYHA II à IV avec dysfonction systolique, chez les patients ayant un rythme sinusal et dont la fréquence cardiaque est ≥ 75 bpm, en association à un traitement conventionnel incluant un traitement par un bêta-bloquant ou si un traitement par un bêta-bloquant est contre-indiqué ou non toléré (voir rubrique 5.1).
04.2 Posologie et mode d'administration
Dosage
Pour les différents dosages, des comprimés pelliculés contenant 5 mg et 7,5 mg d'ivabradine sont disponibles.
Traitement symptomatique de l'angine de poitrine stable chronique
Il est recommandé que la décision d'initier ou d'ajuster le traitement soit prise après des mesures répétées de la fréquence cardiaque, un ECG ou une surveillance ambulatoire de 24 heures.
La dose initiale d'ivabradine ne doit pas dépasser 5 mg deux fois par jour chez les patients de moins de 75 ans. Après 3-4 semaines de traitement, si le patient est toujours symptomatique, si la dose initiale est bien tolérée et si la fréquence cardiaque au repos reste supérieure à 60 bpm, la dose peut être augmentée jusqu'à la dose immédiatement supérieure chez les patients recevant 2,5 mg deux fois par jour ou 5 mg deux fois par jour. La dose d'entretien ne doit pas dépasser 7,5 mg deux fois par jour.
S'il n'y a pas d'amélioration des symptômes de l'angor dans les 3 mois suivant le début du traitement, le traitement par l'ivabradine doit être interrompu.
De plus, s'il n'y a qu'une réponse symptomatique limitée et lorsqu'il n'y a pas de réduction cliniquement pertinente de la fréquence cardiaque au repos dans les trois mois, l'arrêt du traitement doit être envisagé.
Si, pendant le traitement, la fréquence cardiaque au repos diminue en dessous de 50 battements par minute (bpm) ou si le patient signale des symptômes liés à la bradycardie tels que vertiges, fatigue ou hypotension, la posologie doit être ajustée, en tenant également compte de la dose la plus faible de 2,5 mg deux fois par jour (un demi-comprimé de 5 mg deux fois par jour). Après réduction de la dose, la fréquence cardiaque doit être surveillée (voir rubrique 4.4). Le traitement doit être interrompu si la fréquence cardiaque reste inférieure à 50 bpm ou si les symptômes de bradycardie persistent malgré une réduction de la dose.
Traitement de l'insuffisance cardiaque chronique
Le traitement ne doit être instauré que chez les patients présentant une insuffisance cardiaque stable. Il est recommandé que le médecin traitant ait de l'expérience dans le traitement de l'insuffisance cardiaque chronique.
La dose initiale habituelle recommandée d'ivabradine est de 5 mg deux fois par jour. Après deux semaines de traitement, la dose peut être augmentée à 7,5 mg deux fois par jour, si la fréquence cardiaque au repos est continuellement supérieure à 60 bpm, ou diminuée à 2,5 mg deux fois par jour (un demi-comprimé à 5 mg deux fois par jour) si le la fréquence cardiaque au repos reste continuellement inférieure à 50 bpm ou si vous présentez des symptômes liés à une bradycardie tels que des étourdissements, de la fatigue ou une hypotension. Si la fréquence cardiaque est comprise entre 50 et 60 bpm, la dose de 5 mg deux fois par jour doit être maintenue.
Si la fréquence cardiaque au repos diminue de manière persistante en dessous de 50 battements par minute (bpm) pendant le traitement ou si le patient signale des symptômes liés à la bradycardie, la posologie doit être réduite à la dose immédiatement inférieure chez les patients recevant 7, 5 mg deux fois par jour ou 5 mg deux fois par jour . Si la fréquence cardiaque augmente continuellement au-dessus de 60 battements par minute au repos, la dose peut être augmentée à la dose immédiatement supérieure chez les patients prenant 2,5 mg deux fois par jour ou 5 mg deux fois par jour.
Le traitement doit être interrompu si la fréquence cardiaque reste inférieure à 50 bpm ou si les symptômes de bradycardie persistent (voir rubrique 4.4).
Populations particulières
Patients âgés
Chez les patients âgés de 75 ans ou plus, une dose initiale plus faible (2,5 mg deux fois par jour, soit un demi-comprimé à 5 mg deux fois par jour) doit être envisagée avant d'augmenter la dose si nécessaire.
Patients insuffisants rénaux
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale et une clairance de la créatinine supérieure à 15 ml/min (voir rubrique 5.2).
Aucune donnée n'est disponible chez les patients présentant une clairance de la créatinine inférieure à 15 ml/min. L'ivabradine doit donc être utilisée avec prudence chez ce groupe de patients.
Patients souffrant d'insuffisance hépatique
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère. Des précautions doivent être prises lors de la prescription d'ivabradine à des patients présentant une insuffisance hépatique modérée.L'ivabradine est contre-indiquée chez les patients présentant une insuffisance hépatique sévère car elle n'a pas été étudiée dans ce groupe de patients et une augmentation importante de la concentration systémique est attendue (voir rubriques 4.3 et 4.5).
Population pédiatrique
La sécurité et l'efficacité de l'ivabradine dans le traitement de l'insuffisance cardiaque chronique chez les enfants de moins de 18 ans n'ont pas été établies.
Les données disponibles sont décrites dans les rubriques 5.1 et 5.2 mais aucune recommandation sur une posologie ne peut être faite.
Mode d'administration
Les comprimés doivent être pris par voie orale deux fois par jour, c'est-à-dire une fois le matin et une fois le soir, pendant les repas (voir rubrique 5.2).
04.3 Contre-indications
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1
- Fréquence cardiaque au repos inférieure à 70 battements par minute, avant le traitement
- Choc cardiogénique
- Infarctus aigu du myocarde
- Hypotension sévère (
- Insuffisance hépatique sévère
- Syndrome du nœud sinusal
- Bloc sino-auriculaire
- Insuffisance cardiaque aiguë ou instable
- Personnes portant un stimulateur cardiaque (fréquence cardiaque réglée exclusivement par le stimulateur cardiaque)
- Une angine instable
- Bloc AV du troisième degré
- En association avec de puissants inhibiteurs du cytochrome P450 3A4 tels que les antifongiques azolés (kétoconazole, itraconazole), les antibiotiques macrolides (clarithromycine, érythromycine) par os, iosamycine, télithromycine), inhibiteurs de la protéase du VIH (nelfinavir, ritonavir) et néfazodone (voir rubriques 4.5 et 5.2)
- En association avec le vérapamil ou le diltiazem qui sont des inhibiteurs modérés du CYP3A4 avec des propriétés de réduction de la fréquence cardiaque (voir rubrique 4.5)
- Grossesse, allaitement et femmes en âge de procréer n'utilisant pas de mesures contraceptives appropriées (voir rubrique 4.6)
04.4 Mises en garde spéciales et précautions d'emploi appropriées
Avertissements spéciaux
Absence de bénéfice dans les résultats cliniques chez les patients atteints d'angine de poitrine stable chronique symptomatique
L'ivabradine n'est indiquée que pour le traitement symptomatique de l'angine de poitrine stable chronique car l'ivabradine n'a montré aucun bénéfice sur les issues cardiovasculaires (par exemple, infarctus du myocarde ou décès cardiovasculaire) (voir rubrique 5.1).
Mesure de la fréquence cardiaque
Étant donné que la fréquence cardiaque peut fluctuer considérablement au fil du temps, lors de la détermination de la fréquence cardiaque avant le début du traitement par l'ivabradine et lors de l'examen d'un ajustement posologique chez les patients recevant de l'ivabradine, des mesures répétées de la fréquence cardiaque doivent être envisagées. un ECG ou une surveillance ambulatoire de 24 heures. Ce qui précède s'applique également aux patients ayant une fréquence cardiaque basse, en particulier lorsque la fréquence cardiaque diminue en dessous de 50 bpm, ou après une réduction de dose (voir rubrique 4.2).
Arythmies cardiaques
L'ivabradine n'est pas efficace dans le traitement ou la prévention des arythmies cardiaques et est susceptible de perdre son efficacité en cas de tachyarythmie (c'est-à-dire une tachycardie ventriculaire ou supraventriculaire). du nœud sino-auriculaire.
Chez les patients traités par ivabradine, le risque de développer une fibrillation auriculaire est augmenté (voir rubrique 4.8). La fibrillation auriculaire a été rapportée le plus souvent chez les patients prenant de manière concomitante de l'amiodarone ou des antiarythmiques puissants de classe I. Il est recommandé d'effectuer des contrôles cliniques réguliers chez les patients traités par ivabradine pour vérifier l'apparition d'une fibrillation auriculaire (prolongée ou paroxystique). Surveillance ECG, si cliniquement indiquée (par exemple en cas d'aggravation de l'angor, de palpitations, de pouls irrégulier).
Les patients doivent être informés des signes et symptômes de la fibrillation auriculaire et doivent être invités à contacter leur médecin si ces signes et symptômes surviennent.
Si une fibrillation auriculaire se développe pendant le traitement, le rapport bénéfice/risque de la poursuite du traitement par l'ivabradine doit être soigneusement réexaminé.
Les patients atteints d'insuffisance cardiaque chronique présentant des défauts de conduction intraventriculaire (bloc du faisceau gauche, bloc du faisceau droit) et une dyssynchronie ventriculaire doivent être étroitement surveillés.
Utilisation chez les patients présentant un bloc AV du deuxième degré
L'ivabradine n'est pas recommandée chez les patients présentant un bloc AV du deuxième degré.
Utilisation chez les patients ayant une fréquence cardiaque réduite
L'ivabradine ne doit pas être administrée aux patients dont la fréquence cardiaque au repos avant le traitement est inférieure à 70 battements par minute (voir rubrique 4.3).
Si, pendant le traitement, la fréquence cardiaque au repos diminue de manière persistante en dessous de 50 bpm ou si le patient signale des symptômes liés à une bradycardie tels que vertiges, fatigue ou hypotension, la dose doit être réduite ou le traitement doit être arrêté. si la fréquence cardiaque reste inférieure 50 bpm ou si les symptômes dus à une bradycardie persistent (voir rubrique 4.2).
Association avec des inhibiteurs calciques
L'utilisation combinée d'ivabradine avec des inhibiteurs calciques qui réduisent la fréquence cardiaque tels que le vérapamil ou le diltiazem est contre-indiquée (voir rubriques 4.3 et 4.5). L'association d'ivabradine avec des nitrates et des inhibiteurs calciques de type dihydropyridine tels que l'amlodipine n'a suscité aucun problème de sécurité. "L'efficacité supplémentaire" de l'ivabradine en association avec des inhibiteurs calciques de type dihydropyridine n'a pas été démontrée (voir rubrique 5.1).
Insuffisance cardiaque chronique
L'insuffisance cardiaque doit être stable avant d'envisager un traitement par l'ivabradine.L'ivabradine doit être utilisée avec prudence chez les patients atteints d'insuffisance cardiaque de classe fonctionnelle IV de la NYHA, car les données disponibles dans cette population sont limitées.
Accident vasculaire cérébral
L'utilisation d'ivabradine n'est pas recommandée immédiatement après un AVC car aucune donnée n'est disponible.
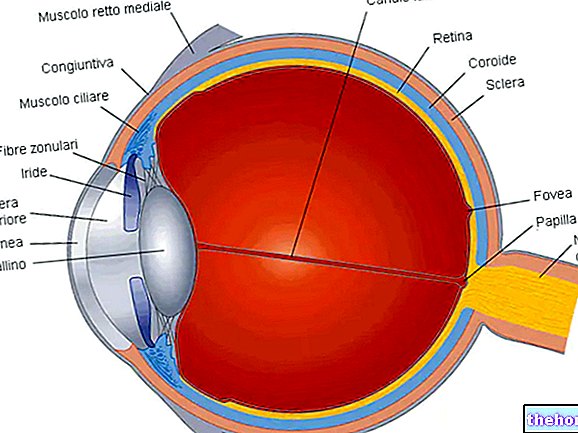
Fonction visuelle
L'ivabradine affecte la fonction rétinienne (voir rubrique 5.1).A ce jour, il n'existe aucune preuve d'un effet toxique de l'ivabradine sur la rétine, cependant les effets sur la fonction rétinienne d'un traitement à plus long terme sont actuellement inconnus jusqu'à un an. L'arrêt du traitement doit être envisagé en cas d'aggravation inattendue de la fonction visuelle.La prudence est de mise chez les patients atteints de rétinite pigmentaire.
Précautions d'emploi
Patients souffrant d'hypotension
Des données limitées sont disponibles chez les patients présentant une hypotension légère à modérée, l'ivabradine doit donc être utilisée avec prudence chez ces patients.L'ivabradine est contre-indiquée chez les patients présentant une hypotension sévère (pression artérielle
Fibrillation auriculaire - Arythmies cardiaques
Il n'y a aucune preuve d'un risque de bradycardie (excessive) lors du retour au rythme sinusal lorsque la cardioversion pharmacologique est entreprise chez les patients recevant de l'ivabradine. Cependant, en l'absence de données complètes, une cardioversion électrique (DC) non urgente doit être envisagée 24 heures après la dernière prise d'ivabradine.
Utilisation chez les patients atteints d'un syndrome du QT long congénital ou traités par des médicaments allongeant l'intervalle QT
L'utilisation d'ivabradine chez les patients atteints d'un syndrome du QT long congénital ou traités par des médicaments allongeant l'intervalle QT doit être évitée (voir rubrique 4.5). Si l'association s'avère nécessaire, une surveillance cardiaque attentive doit être entreprise.
Une fréquence cardiaque réduite, telle que celle causée par l'ivabradine, peut exacerber l'allongement de l'intervalle QT, ce qui peut entraîner de graves arythmies, et en particulier Torse l'orteil.
Patients hypertendus nécessitant des changements de traitement antihypertenseur
Dans l'étude SHIFT, plus de patients ont signalé des épisodes d'augmentation de la pression artérielle sous ivabradine (7,1 %) que de patients sous placebo (6,1 %). Ces épisodes sont survenus plus fréquemment peu de temps après le changement de traitement antihypertenseur, étaient transitoires et n'ont pas affecté l'effet du traitement par l'ivabradine.
Lorsque des modifications du traitement sont apportées chez des patients atteints d'insuffisance cardiaque chronique recevant de l'ivabradine, la pression artérielle doit être surveillée après un intervalle de temps approprié (voir rubrique 4.8).
Excipients
Les comprimés contenant du lactose, les patients présentant des problèmes héréditaires rares d'intolérance au galactose, un déficit en lactase de Lapp ou une malabsorption du glucose et du galactose ne doivent pas prendre ce médicament.
04.5 Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacodynamiques
Combinaisons non recommandées
Médicaments qui prolongent l'intervalle QT
- Médicaments cardiovasculaires qui allongent l'intervalle QT (par exemple quinidine, disopyramide, bépridil, sotalol, ibutilide, amiodarone)
- Médicaments non cardiovasculaires qui allongent l'intervalle QT (par exemple, pimozide, ziprasidone, sertindole, méfloquine, halofantrine, pentamidine, cisapride, érythromycine intraveineuse)
L'utilisation concomitante de médicaments cardiovasculaires et non cardiovasculaires prolongeant l'intervalle QT avec l'ivabradine doit être évitée car l'allongement de l'intervalle QT peut être exacerbé par une diminution de la fréquence cardiaque. Si l'association s'avère nécessaire, une attention particulière doit être accordée à une surveillance cardiaque (voir rubrique 4.4) .
Utilisation concomitante avec précautions
Diurétiques causant une perte de potassium (diurétiques thiazidiques et diurétiques de l'anse) : L'hypokaliémie peut augmenter le risque d'arythmie. Étant donné que l'ivabradine peut provoquer une bradycardie, le résultat de l'association d'une hypokaliémie et d'une bradycardie est un facteur prédisposant aux arythmies sévères, en particulier chez les patients présentant à la fois un syndrome du QT long congénital et d'origine médicamenteuse.
Interactions pharmacocinétiques
Cytochrome P450 3A4 (CYP3A4)
L'ivabradine est métabolisée uniquement par le CYP3A4 et est un très faible inhibiteur de ce cytochrome.Il a été démontré que l'ivabradine n'affecte pas le métabolisme et les concentrations plasmatiques d'autres substrats du CYP3A4 (inhibiteurs faibles, modérés et puissants). Les inhibiteurs et inducteurs du CYP3A4 peuvent interagir avec l'ivabradine et affecter son métabolisme et sa pharmacocinétique à un niveau cliniquement significatif.Des études d'interactions médicamenteuses ont établi que les inhibiteurs du CYP3A4 augmentent les concentrations plasmatiques d'ivabradine, tandis que les inducteurs médicamenteux diminuent. Une augmentation de la concentration plasmatique d'ivabradine peut être associée à un risque de bradycardie excessive (voir rubrique 4.4).
Contre-indications à utiliser en association
L'utilisation concomitante d'inhibiteurs puissants du CYP3A4 tels que les antifongiques azolés (kétoconazole, itraconazole), les antibiotiques macrolides (clarithromycine, érythromycine par os, iosamycine, télithromycine), les inhibiteurs de la protéase du VIH (nelfinavir, ritonavir) et la néfazodone sont contre-indiqués (voir rubrique 4.3). Les puissants inhibiteurs du CYP3A4 kétoconazole (200 mg une fois par jour) et iosamycine (1 g une fois par jour) augmentent la concentration plasmatique moyenne d'ivabradine de 7 à 8 fois.
Inhibiteurs modérés du CYP3A4 : Des études d'interaction spécifiques chez des volontaires sains et des patients ont montré que l'association de l'ivabradine avec des médicaments diminuant la fréquence cardiaque tels que le diltiazem ou le vérapamil entraîne une augmentation de la concentration d'ivabradine (augmentation de l'aire sous la courbe). 2-3 fois) et une diminution supplémentaire de la fréquence cardiaque de 5 bpm. L'utilisation concomitante d'ivabradine avec ces médicaments est contre-indiquée (voir rubrique 4.3).
Utilisation combinée non recommandée
Jus de pamplemousse : la concentration d'ivabradine est doublée après co-administration avec du jus de pamplemousse. Par conséquent, la consommation de jus de pamplemousse doit être évitée.
Précaution d'utilisation en association
- Inhibiteurs modérés du CYP3A4 : L'utilisation de l'ivabradine en association avec d'autres inhibiteurs modérés du CYP3A4 (par exemple le fluconazole) peut être envisagée à la dose initiale de 2,5 mg deux fois par jour et si la fréquence cardiaque au repos est plus élevée à 70 bpm, vérifier la fréquence cardiaque.
- Inducteurs du CYP3A4 : inducteurs du CYP3A4 (par exemple rifampicine, barbituriques, phénytoïne, Hypericum perforatum [millepertuis]) peut diminuer la concentration et l'activité de l'ivabradine. L'utilisation concomitante de médicaments inducteurs du CYP3A4 peut nécessiter un ajustement de la dose d'ivabradine. Il a été démontré que l'utilisation combinée d'ivabradine 10 mg deux fois par jour avec du millepertuis entraîne une réduction de 50 % de l'ASC de l'ivabradine.La prise de millepertuis doit être limitée pendant le traitement par l'ivabradine.
Autres utilisations en association
Des études d'interactions médicamenteuses spécifiques n'ont montré aucun effet cliniquement significatif sur la pharmacocinétique et la pharmacodynamique de l'ivabradine pour les médicaments suivants : inhibiteurs de la pompe à protons (oméprazole, lansoprazole), sildénafil, inhibiteurs de l'HMG CoA réductase (simvastatine), inhibiteurs calciques dihydropyridine (amlopidine, lacipidine ), digoxine et warfarine De plus, il n'y a eu aucun effet cliniquement significatif de l'ivabradine sur la pharmacocinétique de la simvastatine, de l'amlodipine, de la lacidipine, sur la pharmacocinétique et la pharmacodynamique de la digoxine, de la warfarine et sur la pharmacodynamique de l'aspirine.
Pendant les essais cliniques pivot En phase III, les médicaments suivants ont été associés en routine à l'ivabradine sans preuve de sécurité : inhibiteurs de l'enzyme de conversion de l'angiotensine, antagonistes de l'angiotensine II, bêta-bloquants, diurétiques, anti-aldostérone, nitrates de courte et longue durée, inhibiteurs de l'HMG CoA réductase, fibrates, inhibiteurs de la pompe à protons, antidiabétiques oraux, aspirine et autres médicaments antiplaquettaires.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
04.6 Grossesse et allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser des mesures contraceptives appropriées pendant le traitement (voir rubrique 4.3).
Grossesse
Les données sur l'utilisation de l'ivabradine chez la femme enceinte n'existent pas ou sont limitées en nombre. Des études animales ont montré une toxicité pour la reproduction. Ces études ont montré des effets embryotoxiques et tératogènes (voir rubrique 5.3). Le risque potentiel pour l'homme. L'homme est inconnu, par conséquent l'ivabradine est contre-indiquée pendant la grossesse (voir rubrique 4.3).
L'heure du repas
Les études chez l'animal indiquent que l'ivabradine est excrétée dans le lait, c'est pourquoi l'ivabradine est contre-indiquée pendant l'allaitement (voir rubrique 4.3).
Les femmes nécessitant un traitement à l'ivabradine doivent arrêter d'allaiter et choisir une autre méthode d'alimentation pour le bébé.
La fertilité
Les études chez le rat n'ont montré aucun effet sur la fertilité des mâles et des femelles (voir rubrique 5.3).
04.7 Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Une étude spécifique a été menée sur des volontaires sains pour évaluer l'influence possible de l'ivabradine sur les performances de conduite et aucune modification des performances de conduite n'a été retrouvée.Cependant, dans l'expérience post-commercialisation, des cas d'altération de la capacité de conduite ont été rapportés en raison de symptômes visuels. L'ivabradine peut provoquer des phénomènes lumineux transitoires constitués principalement de phosphènes (voir rubrique 4.8). L'apparition éventuelle de ces phénomènes lumineux doit être prise en considération lors de la conduite ou de l'utilisation de machines dans des situations où des changements soudains d'intensité lumineuse peuvent se produire, en particulier lors de la conduite de nuit.
L'ivabradine n'affecte pas l'aptitude à utiliser des machines.
04.8 Effets indésirables
Résumé du profil de sécurité
L'ivabradine a été étudiée dans des essais cliniques impliquant près de 45 000 patients. Les effets indésirables les plus fréquemment observés avec l'ivabradine, phénomènes lumineux (phosphènes) et bradycardie, sont dose-dépendants et corrélés à l'effet pharmacologique du médicament.
Tableau des effets indésirables
Les effets indésirables suivants ont été observés au cours des essais cliniques et sont répertoriés selon la fréquence suivante : très fréquent (≥1/10) ; commun (≥1 / 100,
* Fréquence calculée à partir d'essais cliniques pour les événements indésirables signalés à partir de rapports spontanés
Description des effets indésirables sélectionnés
Des phénomènes lumineux (phosphènes) ont été rapportés par 14,5 % des patients, décrits comme une « augmentation transitoire de la luminosité dans une » zone limitée du champ visuel. Ils sont généralement déclenchés par des changements soudains d'intensité lumineuse.Les phosphènes peuvent également être décrits comme un halo, une décomposition d'images (effets stroboscopiques ou kaléidoscopiques), des lumières colorées intenses ou des images multiples (persistance rétinienne). L'apparition des phosphènes survient généralement au cours des deux premiers mois de traitement, après quoi ils peuvent survenir à plusieurs reprises.
Les phosphènes sont généralement signalés comme étant d'intensité légère ou modérée. Tous les phosphènes se sont résolus pendant ou après le traitement et la majorité (77,5 %) se sont résolus pendant le traitement. Moins de 1 % des patients ont modifié leurs habitudes quotidiennes ou ont dû arrêter leur traitement à cause des phosphènes.
Une bradycardie a été rapportée par 3,3% des patients, principalement au cours des 2-3 premiers mois après le début du traitement. 0,5% des patients ont présenté une bradycardie sévère avec une fréquence cardiaque inférieure ou égale à 40 bpm.
Dans l'étude SIGNIFY, une fibrillation auriculaire a été observée chez 5,3 % des patients sous ivabradine contre 3,8 % des patients du groupe placebo. Dans un analyse groupée de tous les essais cliniques contrôlés en double aveugle de phase II/III d'une durée d'au moins trois mois, qui ont inclus plus de 40 000 patients, l'incidence de la fibrillation auriculaire était de 4,86 % chez les patients traités par l'ivabradine, contre 4,08 % dans le groupe témoin, qui correspond à un hazard ratio de 1,26, IC 95% [1,15 - 1,39].
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés survenant après l'autorisation du médicament est importante car elle permet un suivi continu du rapport bénéfice/risque du médicament.Les professionnels de santé sont invités à déclarer tout effet indésirable suspecté via le site internet : www. agenziafarmaco .gov.it / it / managers de l'Agence italienne des médicaments.
04.9 Surdosage
Symptômes
Un surdosage peut entraîner une bradycardie sévère et prolongée (voir rubrique 4.8).
La gestion
La bradycardie sévère doit être traitée de manière symptomatique dans un cadre spécialisé. En cas de bradycardie avec une mauvaise tolérance hémodynamique, un traitement symptomatique comprenant l'utilisation intraveineuse de bêta-agonistes tels que l'isoprénaline peut être envisagé. Si nécessaire, une électrostimulation cardiaque temporaire peut être instaurée.
05.0 PROPRIÉTÉS PHARMACOLOGIQUES
05.1 Propriétés pharmacodynamiques
Classe pharmacothérapeutique : thérapie cardiaque.
Code ATC : C01EB17.
Mécanisme d'action
L'ivabradine est un médicament qui réduit sélectivement la fréquence cardiaque en agissant par inhibition sélective et spécifique du courant du stimulateur cardiaque LES f, qui contrôle la dépolarisation diastolique spontanée dans le nœud sinusal et régule la fréquence cardiaque. Les effets cardiaques sont spécifiques du nœud sinusal sans effet sur les temps de conduction intra-auriculaire, auriculo-ventriculaire ou intraventriculaire, ni sur la contractilité myocardique ou la repolarisation ventriculaire.
L'ivabradine peut également interagir avec le courant LES h présent dans la rétine et qui a des caractéristiques très proches de celle du courant cardiaque LES F. Ce courant intervient dans le processus de résolution temporelle du système visuel, réduisant la réponse rétinienne aux stimuli lumineux intenses. Dans certaines circonstances de déclenchement (par exemple des changements rapides de luminosité), une inhibition partielle de LES h de la part de l'ivabradine sous-tend les phénomènes lumineux qui peuvent occasionnellement être rapportés par les patients.Les phénomènes lumineux (phosphènes) sont décrits comme une « augmentation transitoire de la luminosité dans une » zone limitée du champ visuel (voir rubrique 4.8).
Effets pharmacodynamiques
La principale propriété pharmacodynamique de l'ivabradine chez l'homme est une réduction spécifique dose-dépendante de la fréquence cardiaque. L'analyse de la réduction de la fréquence cardiaque avec des doses allant jusqu'à 20 mg deux fois par jour indique qu'il existe une tendance à atteindre un plateau, ce qui est cohérent avec le risque réduit d'avoir des bradycardies sévères avec une fréquence inférieure à 40 bpm (voir rubrique 4.8).
Aux doses normalement recommandées, la réduction de la fréquence cardiaque est d'environ 10 bpm au repos et pendant l'exercice. Cela conduit à une réduction de la charge de travail cardiaque et de la consommation d'oxygène par le myocarde. L'ivabradine n'affecte pas la conduction intracardiaque, la contractilité (absence d'effet inotrope négatif) ou la repolarisation ventriculaire :
- dans les études cliniques électrophysiologiques, l'ivabradine n'a eu aucun effet sur les temps de conduction auriculo-ventriculaire ou intraventriculaire ou sur l'intervalle QT corrigé ;
- chez les patients présentant une dysfonction ventriculaire gauche (fraction d'éjection ventriculaire gauche (FEVG) comprise entre 30 et 45 %), l'ivabradine n'a pas eu d'effet indésirable sur la fraction d'éjection.
Efficacité et sécurité cliniques
L'efficacité antiangineuse et anti-ischémique de l'ivabradine a été évaluée dans cinq essais cliniques randomisés en double aveugle (trois versus placebo, et les autres versus aténolol et amlodipine, respectivement).Ces études ont inclus un total de 4 111 patients atteints d'angine de poitrine chronique stable , dont 2 617 traités à l'ivabradine.
L'ivabradine 5 mg deux fois par jour s'est avérée efficace sur les paramètres de l'épreuve d'effort dans les 3 à 4 semaines suivant le traitement. L'efficacité a été confirmée avec 7,5 mg deux fois par jour. En particulier, le bénéfice supplémentaire par rapport à la dose de 5 mg deux fois par jour a été établi dans une étude contrôlée par rapport à l'aténolol : la durée totale d'exercice évaluée à la valeur minimale d'efficacité a été augmentée d'environ 1 minute après un mois de traitement par 5 mg deux fois. quotidiennement et encore amélioré de près de 25 secondes après une période ultérieure de 3 mois de titration forcée à 7,5 mg deux fois par jour. L'efficacité de 5 et 7,5 mg deux fois par jour sur les paramètres du test ergométrique s'est avérée cohérente dans toutes les études (durée totale de l'exercice, temps jusqu'à l'arrêt du test d'effort de douleur angineuse, temps d'apparition de la douleur angineuse et temps d'apparition de la 1 mm de déplacement du segment ST) et a été associée à une diminution d'environ 70 % de la fréquence des crises d'angine. Le régime biquotidien a donné une « efficacité uniforme » sur une période de 24 heures.
Dans une étude randomisée contrôlée contre placebo chez 889 patients, l'ivabradine administrée en plus de l'aténolol 50 mg une fois par jour a démontré une efficacité additive sur tous les paramètres du test d'effort (ETT) au creux de l'activité du médicament (12 heures après la prise orale).
Dans une étude randomisée contrôlée contre placebo chez 725 patients, l'ivabradine n'a pas montré d'efficacité additive en plus de l'amlodipine 10 mg une fois par jour au creux de l'activité du médicament (12 heures après la prise orale), alors qu'une efficacité additive a été démontrée au pic (3- 4 heures après la prise orale).
Dans une étude randomisée contrôlée contre placebo chez 1 277 patients, l'ivabradine a démontré une efficacité additive statistiquement significative sur la réponse au traitement (définie comme une réduction d'au moins 3 crises d'angor par semaine et/ou un allongement d'au moins 60 secondes du temps jusqu'au sous-niveau). mm du segment ST lors du stress test al tapis roulant) en plus de l'amlodipine 5 mg une fois par jour ou de la nifédipine GITS 30 mg une fois par jour, au minimum d'activité médicamenteuse (12 heures après la prise d'ivabradine orale) pendant une période de traitement de 6 semaines (OR = 1, 3, IC 95% [1,0 -1,7] ; p = 0,012). L'ivabradine n'a pas montré d'efficacité additive sur les autres paramètres du test d'effort (critères secondaires) à l'activité minimale du médicament, alors qu'elle a montré une efficacité additive au pic d'activité (3-4 heures après la prise d'ivabradine par voie orale).
L'efficacité de l'ivabradine a été entièrement maintenue pendant les périodes de traitement de 3 ou 4 mois dans les études d'efficacité clinique. Il n'y avait aucune preuve de développement d'une tolérance au médicament (perte d'efficacité) pendant le traitement, ni de phénomène rebond après l'arrêt brutal du traitement. Les effets anti-angineux et anti-ischémiques de l'ivabradine ont été associés à une réduction dose-dépendante de la fréquence cardiaque et à une réduction significative du produit fréquence-pression (fréquence cardiaque x pression artérielle systolique) au repos et pendant l'exercice. Les effets sur la pression artérielle et la résistance vasculaire périphérique étaient mineurs et cliniquement insignifiants.
Une réduction soutenue de la fréquence cardiaque a été démontrée chez les patients traités par ivabradine pendant au moins un an (n = 713). Aucune influence sur le métabolisme des lipides ou des glucides n'a été observée.
L'efficacité antiangineuse et anti-ischémique de l'ivabradine est également maintenue chez les patients diabétiques (n = 457) avec un profil de tolérance similaire à celui observé dans la population générale.
Une vaste étude de résultats, BEAUTIFUL, a été menée chez 10917 patients atteints de maladie coronarienne et de dysfonctionnement ventriculaire gauche (infarctus aigu du myocarde FEVG ou hospitalisation pour insuffisance cardiaque d'apparition ou d'aggravation. L'étude n'a montré aucune différence dans le taux de résultat principal composite dans l'ivabradine. groupe versus groupe placebo (risque relatif ivabradine : placebo 1,00, p = 0,945).
Dans l'analyse post-hoc d'un sous-groupe de patients présentant un angor symptomatique lors de la randomisation (n = 1507), il n'y a eu aucun rapport de tolérance de décès cardiovasculaire, d'hospitalisation pour infarctus aigu du myocarde ou d'insuffisance cardiaque (ivabradine 12, 0 % versus placebo 15,5 %, p = 0,05).
Une vaste étude de résultats cliniques, SIGNIFY, a été menée chez 19 102 patients atteints de maladie coronarienne et sans insuffisance cardiaque cliniquement évidente (FEVG> 40 %), en plus d'un traitement de fond optimal. Un régime plus élevé que la posologie approuvée a été utilisé (dose initiale de 7,5 mg deux fois par jour (5 mg deux fois par jour, si âge ≥ 75 ans) et titré jusqu'à 10 mg deux fois par jour. ). Le critère principal d'efficacité était le composite décès cardiovasculaire ou infarctus du myocarde non mortel. L'étude n'a montré aucune différence sur la fréquence du critère d'évaluation principal composite (PCE) dans le groupe ivabradine versus le groupe placebo (risque relatif ivabradine/placebo 1,08, p = 0,197). Une bradycardie a été rapportée chez 17,9 % des patients du groupe ivabradine ( 2,1 % dans le groupe placebo) 7,1 % des patients ont reçu du vérapamil, du diltiazem ou des inhibiteurs puissants du CYP3A4 au cours de l'étude.
Une petite augmentation statistiquement significative de la PCE a été observée dans un sous-groupe prédéfini de patients souffrant d'angor à l'inclusion, de classe II ou supérieure de la SCC (n = 12 049) (taux annuels de 3,4 % contre 2,9 %, risque relatif ivabradine/placebo de 1,18, p = 0,018) , mais pas dans le sous-groupe de la population totale de patients angineux dans la classe CCS ≥ I (n = 14 286) (risque relatif ivabradine / placebo 1,11, p = 0,110).
La dose utilisée dans l'étude, supérieure à celle approuvée, n'expliquait pas complètement les résultats obtenus.
L'étude SHIFT est une vaste étude multicentrique, internationale, randomisée, contrôlée, en double aveugle, contrôlée par placebo, menée auprès de 6 505 patients adultes atteints d'insuffisance cardiaque chronique (de ≥4 semaines), NYHA classe II à IV, avec une fraction d'éjection ventriculaire gauche réduite (FEVG ≤ 35%) et une fréquence cardiaque au repos 70 bpm.
Les patients ont reçu un traitement conventionnel comprenant des bêtabloquants (89 %), des inhibiteurs de l'ECA et/ou des antagonistes de l'angiotensine II (91 %), des diurétiques (83 %) et des agents anti-aldostérone (60 %). % de patients ont été traités avec 7,5 mg deux fois par jour. Le suivi médian était de 22,9 mois. Le traitement par ivabradine a été associé à une réduction moyenne de la fréquence cardiaque de 15 bpm par rapport à 80 bpm au départ La différence de fréquence cardiaque entre le bras ivabradine et le placebo était de 10,8 bpm à 28 jours, de 9,1 bpm à 12 mois et de 8,3 bpm à 24 mois.
L'étude a démontré une réduction cliniquement et statistiquement significative de 18 % du risque relatif de la fréquence du critère principal composite de mortalité cardiovasculaire et d'hospitalisation pour aggravation de l'insuffisance cardiaque (hazard ratio : 0,82, IC à 95 % [0,75 ; 0,90] - p
Effet du traitement sur le critère principal composite, ses composantes et critères secondaires
La réduction observée du critère d'évaluation principal a été maintenue quel que soit le sexe, la classification NYHA, l'étiologie ischémique ou non ischémique de l'insuffisance cardiaque et les antécédents de diabète ou d'hypertension.
Dans le sous-groupe de patients atteints de mucoviscidose ≥ 75 bpm (n = 4 150), une réduction plus importante du critère principal composite de 24 % a été observée (hazard ratio : 0,76, IC à 95 % [0,68 ; 0,85] -p
Dans ce sous-groupe de patients, le profil de tolérance de l'ivabradine est cohérent avec celui de la population totale.
Un effet significatif sur le critère principal composite a été observé dans l'ensemble du groupe de patients recevant un traitement bêtabloquant (hazard ratio : 0,85, IC 95% [0,76 ; 0,94]).
Dans le sous-groupe de patients atteints de mucoviscidose ≥ 75 bpm et à la dose optimale recommandée de bêta-bloquant, aucun bénéfice statistiquement significatif n'a été observé sur le critère principal composite (hazard ratio : 0,97, IC à 95 % [0,74 ; 1,28]) et sur les autres critères secondaires, y compris l'hospitalisation pour aggravation de l'insuffisance cardiaque (rapport de risque : 0,79, IC à 95 % [0,56 ; 1,10]) ou décès par insuffisance cardiaque (rapport de risque : 0,69, IC à 95 % [0,31 ; 1,53]).
Une amélioration significative de la classe NYHA a été rapportée lors de la dernière enquête : elle s'est améliorée chez 887 patients (28 %) traités par ivabradine contre 776 patients (24 %) traités par placebo (p = 0,001).
Population pédiatrique
Une étude randomisée, en double aveugle, contrôlée contre placebo a été réalisée chez 116 patients pédiatriques (17 âgés de 6 à 12 mois, 36 âgés de 1 à 3 ans et 63 âgés de 3 à 18 ans) atteints d'insuffisance cardiaque chronique et de cardiomyopathie dilatée (DCM) en plus du traitement de base optimal. 74 patients ont reçu de l'ivabradine (avec un ratio de 2:1). La dose initiale était de 0,02 mg/kg deux fois par jour dans la tranche d'âge de 6 à 12 mois, de 0,05 mg/kg deux fois par jour dans la tranche d'âge de 1 à 3 ans et dans la tranche d'âge de 1 à 3 ans 3 et 18 ans avec le poids corporel poids corporel 40 kg. La dose a été ajustée en fonction de la réponse thérapeutique avec une dose maximale de 0,2 mg/kg deux fois par jour, 0,3 mg/kg deux fois par jour et 15 mg/kg deux fois par jour, respectivement. Dans cette étude, l'ivabradine a été administrée sous forme de formulation liquide orale ou sous forme de comprimé deux fois par jour. L'absence de différences pharmacocinétiques entre les 2 formulations a été démontrée dans une étude croisée en ouvert, randomisée, sur deux périodes menée chez 24 volontaires adultes sains.
Une réduction de 20 % de la fréquence cardiaque, sans bradycardie, a été obtenue chez 69,9 % des patients du groupe ivabradine versus 12,2 % dans le groupe placebo pendant la période de titration de 2 à 8 semaines (rapport de cotes : E = 17,24, IC à 95 % [ 5,91 ; 50,30]).
La dose moyenne d'ivabradine qui a entraîné une réduction de 20 % de la fréquence cardiaque était respectivement de 0,13 ± 0,04 mg/kg deux fois par jour, 0,10 ± 0,04 mg/kg deux fois par jour et 4,1 ± 2,2 mg deux fois par jour dans les sous-groupes d'âge 1 à 3 ans. , 3 à 18 ans et poids corporel
Après 12 mois de traitement, la fraction d'éjection ventriculaire gauche moyenne est passée de 31,8 % à 45,3 % dans le groupe ivabradine contre une augmentation de 35,4 % à 42,3 % dans le groupe placebo. C" était une amélioration de la classe NYHA chez 37,7 % des patients traités par l'ivabradine par rapport à 25,0 % des patients du groupe placebo. Ces améliorations n'étaient pas statistiquement significatives.
Le profil de tolérance sur un an était similaire à celui décrit chez les patients adultes atteints d'insuffisance cardiaque chronique.
Les effets à long terme de l'ivabradine sur la croissance, la puberté et le développement général ainsi que l'efficacité à long terme du traitement par l'ivabradine pendant l'enfance pour réduire les maladies cardiovasculaires/la mortalité n'ont pas été étudiés.
L'Agence européenne des médicaments a renoncé à l'obligation de soumettre les résultats des études avec Procoralan dans tous les sous-ensembles de la population pédiatrique pour le traitement de l'angine de poitrine.
L'Agence européenne des médicaments a renoncé à l'obligation de soumettre les résultats des études avec Procoralan chez les enfants de moins de 6 mois dans le traitement de l'insuffisance cardiaque chronique.
05.2 Propriétés pharmacocinétiques
Dans des conditions physiologiques, l'ivabradine est rapidement libérée des comprimés et est très soluble dans l'eau (> 10 mg/ml).L'ivabradine est l'énantiomère S et aucune bioconversion n'a été démontrée. in vivo. Le dérivé N-déméthylé de l'ivabradine a été identifié comme le principal métabolite actif chez l'homme.
Absorption et biodisponibilité
L'ivabradine est rapidement et presque complètement absorbée après administration orale, avec un pic plasmatique atteint en une heure environ à jeun. La biodisponibilité absolue des comprimés pelliculés est d'environ 40 %, en raison de l'effet de premier passage dans l'intestin et le foie.
La nourriture retarde l'absorption d'environ une heure et augmente sa présence dans le plasma de 20 à 30 %. Il est recommandé de prendre le comprimé avec les repas pour diminuer la variabilité intra-individuelle de la concentration (voir rubrique 4.2).
Distribution
L'ivabradine est liée à environ 70 % aux protéines plasmatiques et, chez les patients, le volume de distribution à l'état d'équilibre est proche de 100 L. La concentration plasmatique maximale après administration chronique à la dose recommandée de 5 mg deux fois par jour est de 22 ng/mL (CV = 29 %. La concentration plasmatique moyenne à l'état d'équilibre est de 10 ng/mL (CV = 38 %).
Biotransformation
L'ivabradine est largement métabolisée par le foie et l'intestin par des oxydations catalysées uniquement par le cytochrome P450 3A4 (CYP3A4). Le principal métabolite actif est le dérivé N-déméthylé (S18982), avec une concentration d'environ 40 % de celle de la molécule mère. Le métabolisme de ce métabolite actif implique également le CYP3A4. L'ivabradine a une faible affinité pour le CYP3A4, ne montre aucune induction ou inhibition cliniquement significative du CYP3A4 et est donc peu susceptible de modifier le métabolisme ou les concentrations plasmatiques des substrats du CYP3A4. En revanche, les inhibiteurs et inducteurs puissants peuvent modifier substantiellement les concentrations plasmatiques. ivabradine (voir rubrique 4.5 ).
Élimination
L'ivabradine est éliminée avec une demi-vie principale de 2 heures (70-75% de l'ASC) dans le plasma et une demi-vie effective de 11 heures. La clairance totale est d'environ 400 ml/min et la clairance rénale est d'environ 70 ml/min. L'excrétion des métabolites se produit à parts égales avec les fèces et l'urine. Environ 4 % d'une dose orale sont excrétés sous forme inchangée dans les urines.
Linéarité / Non Linéarité
La cinétique de l'ivabradine est linéaire sur la plage de doses orales de 0,5 à 24 mg.
Populations particulières
- Sujets âgés : Aucune différence pharmacocinétique (ASC et Cmax) n'a été observée entre les patients âgés (≥ 65 ans) ou très âgés (≥ 75 ans) et la population générale (voir rubrique 4.2).
- Insuffisance rénale : l'impact de l'insuffisance rénale (clairance de la créatinine 15 à 60 ml/min) sur la pharmacocinétique de l'ivabradine est minime, en accord avec la contribution modeste de la clairance rénale (environ 20 %) à l'excrétion totale de l'ivabradine et de son métabolite majeur S18982 ( voir rubrique 4.2).
- Insuffisance hépatique : Chez les patients présentant une insuffisance hépatique légère (score de Child Pugh jusqu'à 7), l'ASC de l'ivabradine libre et de son principal métabolite actif est environ 20 % plus élevée que chez les sujets ayant une fonction hépatique normale. Les données sont insuffisantes pour tirer des conclusions chez les patients présentant une insuffisance hépatique modérée. Il n'y a pas de données disponibles chez les patients présentant une insuffisance hépatique sévère (voir rubriques 4.2 et 4.3).
- Population pédiatrique : Le profil pharmacocinétique de l'ivabradine chez les patients pédiatriques atteints d'insuffisance cardiaque chronique âgés de 6 mois à 18 ans est similaire au profil pharmacocinétique décrit chez l'adulte lors de l'application d'un schéma de titration basé sur l'âge et le poids.
Relation pharmacocinétique / pharmacodynamique (PK / PD)
L'analyse de la relation PK/PD a montré que la fréquence cardiaque diminue pratiquement linéairement avec l'augmentation des concentrations plasmatiques d'ivabradine et de S18982 pour des doses allant jusqu'à 15-20 mg deux fois par jour. A des doses plus élevées, la diminution de la fréquence cardiaque n'est plus proportionnelle aux concentrations plasmatiques d'ivabradine et tend à atteindre une plateau. Des concentrations élevées d'ivabradine, qui peuvent survenir lorsque l'ivabradine est co-administrée avec des inhibiteurs puissants du CYP3A4, peuvent entraîner une diminution excessive de la fréquence cardiaque bien que ce risque soit réduit avec des inhibiteurs modérés du CYP3A4 (voir rubriques 4.3, 4.4 et 4.5). La relation PK/PD de l'ivabradine chez les patients pédiatriques atteints d'insuffisance cardiaque chronique âgés de 6 mois à 18 ans est similaire à celle décrite chez l'adulte.
05.3 Données de sécurité précliniques
Les données non cliniques ne révèlent aucun risque particulier pour l'homme sur la base des études conventionnelles de sécurité pharmacologie, toxicité à doses répétées, génotoxicité, potentiel cancérigène. Des études de toxicité sur la reproduction ont montré que l'ivabradine n'avait aucun effet sur la fertilité des rats mâles et femelles.Lorsque des animaux gravides ont été traités au cours de l'organogenèse avec des doses proches de celles thérapeutiques, une incidence plus élevée de fœtus présentant des malformations a été observée chez le rat et un petit nombre de foetus avec ectrodactylie chez le lapin.
Chez les chiens traités par l'ivabradine (doses de 2, 7 ou 24 mg/kg/jour) pendant un an, des modifications réversibles de la fonction rétinienne ont été observées mais n'ont pas été associées à des lésions des structures oculaires. Ces données sont cohérentes avec les effets pharmacologiques de l'ivabradine et sont attribuables à son interaction avec les LES h activé en hyperpolarisation, présent dans la rétine, et qui partage une large homologie avec le courant du stimulateur cardiaque LES F.
D'autres études à long terme à doses répétées et études de cancérogénicité n'ont révélé aucun changement cliniquement pertinent.
Évaluation des risques environnementaux (Évaluation des risques environnementaux, ÉTAIT)
L'évaluation des risques environnementaux de l'ivabradine a été réalisée conformément aux directives européennes de l'ERA.
Les résultats de ces évaluations confirment l'absence de risque environnemental de l'ivabradine et l'ivabradine ne pose pas de danger pour l'environnement.
06.0 INFORMATIONS PHARMACEUTIQUES
06.1 Excipients
Noyau
Lactose monohydraté
Stéarate de magnésium (E470B)
Fécule de maïs
Maltodextrine
Silice colloïdale anhydre (E551)
Film de revêtement
Hypromellose (E464)
Dioxyde de titane (E171)
Macrogol 6000
Glycérol (E422)
Stéarate de magnésium (E470B)
Oxyde de fer jaune (E172)
Oxyde de fer rouge (E172)
06.2 Incompatibilité
Non pertinent.
06.3 Durée de validité
3 années.
06.4 Précautions particulières de conservation
Ce médicament ne nécessite aucune condition particulière de conservation.
06.5 Nature du conditionnement primaire et contenu de l'emballage
Blister aluminium/PVC contenu dans des boîtes en carton.
Packs
Boîtes calendrier contenant 14, 28, 56, 84, 98, 100 ou 112 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
06.6 Instructions d'utilisation et de manipulation
Pas d'instructions particulières.
07.0 TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHE
Les Laboratoires Servier
50, rue Carnot
92284 Suresnes cedex
La France
08.0 NUMÉRO D'AUTORISATION DE MISE SUR LE MARCHÉ
UE / 1/05/316/001 - 007
037061013
037061025
037061049
037061052
037061064
037061076
A.I.C. N° 037061037/E : Procoralan 5 mg comprimés pelliculés - boîte de 56 comprimés
09.0 DATE DE PREMIÈRE AUTORISATION OU DE RENOUVELLEMENT DE L'AUTORISATION
Date de première autorisation : 25/10/2005
Date du dernier renouvellement : 25/10/2010
10.0 DATE DE RÉVISION DU TEXTE
03/2015